Proquest Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structural Basis for the Coupling Mechanism of Respiratory Complex I, a Giant Molecular Proton Pump
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector e4 Abstracts opening a large membrane pore, potentiated by inducers such as the P3 & P4.3 phosphate, the depletion of adenine nucleotides and the presence of thiol oxidants. It leads to the swelling of the mitochondria, loss of Electron cryomicroscopy investigation of the proton-conducting proton motive force, disruption of ion homeostasis and hydrolysis of pore of ATP synthase ATP by the ATP synthase. These events have been linked to pathways John Rubinstein leading to cell death, and to human diseases including cardiac The Hospital for Sick Children Research Institute, 686 Bay Street, ischemia and muscle dystrophy. We have examined this proposal, Rm. 20-9705, Toronto, ON, Canada fi and a summary of our ndings will be presented. E-mail: [email protected] Adenosine triphosphate (ATP) synthases use the energy stored in doi:10.1016/j.bbabio.2014.05.120 a transmembrane proton-motive force to synthesize ATP from aden- osine diphosphate (ADP) and inorganic phosphate. The transloca- tion of protons across the membrane region of the complex leads to P3 & P4.2 rotation of a rotor subcomplex that drives conformational changes in the enzyme's membrane-extrinsic catalytic region. The mechanism by Structural basis for the coupling mechanism of respiratory which proton translocation induces rotation is not known. Here, we complex I, a giant molecular proton pump present structural data from single particle electron cryomicroscopy Leonid Sazanov (cryo-EM) that gives insight into the architecture of the membrane- Mitochondrial Biology Unit, Medical Research Council, Cambridge, UK bound motors of ATP synthases and related enzymes. -

5Th ISMB Retreat, 3-4 July 2013
5th ISMB Retreat 3 – 4 July 2013 Report by Dr. Clare Sansom, Department of Biological Sciences, Birkbeck The Institute of Structural Molecular Biology 2001, Cambridge. He has received numerous (ISMB), set up to foster collaboration honours and awards and is a Fellow of the between researchers in University College Royal Society and a Foreign Member of the London and Birkbeck who work in molecular National Academy of Sciences of the US. and structural biology and allied disciplines, is now ten years old. It has established two Dobson’s research has been particularly series of biannual research meetings focused on the mechanisms through which designed to grow these collaborations, proteins fold, or on some occasions misfold. inspire research excellence and encourage This area of science has become increasingly new ideas. The ISMB symposia, held in even important because of its link with human years, feature lectures by world-class disorders and strategies through which they researchers in its constituent disciplines, and can be combatted. Dobson’s recent work is the residential retreats, held in odd years, summed up in the title of his talk: ‘New are dominated by talks by postgraduate Approaches to Understanding and Preventing students and young postdoctoral researchers Neurodegenerative Diseases’. He began with from its core departments. The retreat held a discussion of this group of diseases, on July 3 and 4 2013 was therefore the fifth explaining how the prevalence of in the series. It was also the third to be held neurodegenerative disorders, and in the quiet surroundings of Robinson College Alzheimer’s disease in particular, is in Cambridge. -

Why Are Cells Powered by Proton Gradients? By: Nick Lane, Ph.D
Proton Gradient, Cell Origin, ATP Synthase | Lear... http://www.nature.com/scitable/topicpage/why-are... CELL ORIGINS AND METABOLISM | Lead Editor: Gary Coté, Mario De Tullio Why Are Cells Powered by Proton Gradients? By: Nick Lane, Ph.D. (Research Department of Genetics, Evolution and Environment, University College London ) © 2010 Nature Education Citation: Lane, N. (2010) Why Are Cells Powered by Proton Gradients? Nature Education 3(9):18 The proton gradients that power respiration are as universal as the genetic code itself, giving an insight into the origin of life and the singular origin of complexity Why do virtually all cells "breathe" by pumping protons (hydrogen ions) across a membrane? According to molecular biologist Leslie Orgel, this is the single most counterintuitive idea in biology after Darwin's, and the only one to bear comparison with the concepts of Heisenberg, Schrödinger, and Einstein (Orgel 1999). Pioneered by the eccentric British biochemist Peter Mitchell (Figure 1), largely in his own research laboratories in a renovated country house in rural Cornwall, the concept was controversial for more than twenty years. This period of controversy was known as the "ox-phos wars" (after "oxidative phosphorylation," the mechanism of ATP synthesis in respiration). The wars drew to an end only after Mitchell received the Nobel Prize in 1978. There's an irony here. Mitchell's Nobel was for work in chemistry, yet his Figure 1: Peter Mitchell and the ATP synthase enzyme ideas are actually about the elimination of chemistry. In the same way © 1999 Nature Publishing Group Orgel, L. Are you serious, Dr that the genetic code enables information to transcend chemistry, so Mitchell? Nature 402, 17 (1999). -

PRESS RELEASE Embargoed Until 1800 London Time / 1300 US
PRESS RELEASE August 27, 2019 Embargoed until 1800 London time / 1300 US Eastern Time on 28 August 2019 High-end microscopy reveals structure and function of crucial metabolic enzyme Structural biologists reveal the atomic structure and regulative mechanism of the metabolic enzyme transhydrogenase – Results are the first to be published based on state-of-the-art cryo-electron microscope at IST Austria The enzyme transhydrogenase plays a central role in regulating metabolic processes in animals and humans alike. Malfunction can lead to serious disorders. For the first time, structural biologists at the Institute of Science and Technology Austria (IST Austria) have now visualized and analyzed the enzyme’s atomic structure with the support of the institute’s newly installed high- end cryo-electron microscope. The data presented in the journal Nature are relevant for the development of currently unavailable therapeutic options. Within each cell, the power houses called mitochondria continuously break down molecules derived from food to generate energy as well as to produce new molecules that serve as building blocks of cells. Balancing these two opposing processes is accomplished by an enzyme called proton- translocating transhydrogenase or NNT (nicotinamide nucleotide transhydrogenase). NNT sits in the mitochondria’s membrane and uses the electrochemical proton gradient generated by cellular respiration to provide the mitochondria with just the right amount of the co-enzyme NADPH, a vital metabolic precursor. The proper functioning of NNT is crucial for metabolic regulation in all animals including humans. However, the details of how NNT accomplishes the coordinated transfer of protons across the membrane and synthesis of NADPH have remained obscure due to the lack of knowledge about the enzyme’s atomic structure. -

Pagina 1 Di 304 SBBL - SPRINGER E-BOOKS : TITOLI CORRENTI 2014 N
SBBL - SPRINGER E-BOOKS : TITOLI CORRENTI 2014 N. Book Title Author Year Editore n. 1 (Endo)symbiotic Methanogenic Archaea Johannes H.P. Hackstein 2010 Springer Berlin Heidelberg n. 2 20 Years of Computational Neuroscience Tim R. New 2011 Springer Netherlands n. 3 25 Years of p53 Research James M Bower 2013 Springer New York n. 4 34th Hemophilia Symposium Pierre Hainaut, Klas G. Wiman 2005 Springer Netherlands n. 5 35th Hemophilia Symposium I. Scharrer, W. Schramm 2005 Springer Berlin Heidelberg n. 6 36th Hemophilia Symposium Hamburg 2005 Inge Scharrer, Wolfgang Schramm 2006 Springer Berlin Heidelberg n. 7 37th Hemophilia Symposium Inge Scharrer, Wolfgang Schramm 2007 Springer Berlin Heidelberg n. 8 3D Histology Evaluation of Dermatologic Surgery Inge Scharrer, Wolfgang Schramm 2008 Springer Berlin Heidelberg n. 9 41es Journées nationales de la Société Française de Médecine Périnatale (Grenoble 12–14 octobre 2011) Helmut Breuninger, Patrick Adam 2013 Springer London n. 10 42es Journées nationales de la Société Française de Médecine Périnatale (Montpellier 17–19 octobre 2012) Société Française de Médecine Périnatale 2011 Springer Paris n. 11 50 Years of Phytochemistry Research Société Française de Médecine Périnatale 2013 Springer Paris n. 12 55 Years German Society of Anaesthesiology and Intensive Care Medicine David R. Gang 2013 Springer International Publishing n. 13 5-HT2C Receptors in the Pathophysiology of CNS Disease J. Schüttler 2012 Springer Berlin Heidelberg n. 14 60 Years of Survival Outcomes at The University of Texas MD Anderson Cancer Center Giuseppe Di Giovanni, Ennio Esposito, Vincenzo Di Matteo 2011 Humana Press n. 15 7.0 Tesla MRI Brain Atlas M. -

The Evolving Concept of Mitochondria
The Evolving Concept of Mitochondria: Cold Spring Harbor Laboratory | Meetings & Courses Program From Symbiotic Origins to Therapeutic Opportunities October 18 - 21, 2018 Poster Abstract Deadline: September 15 Meeting Website: meetings.cshl.edu/history18 Cold Spring Harbor, New York Mary Herbert, Newcastle University, UK Organizers Henry Higgs, Dartmouth Medical School Anu Suomalainen**, University of Helsinki, Finland Judy Hirst, MRC Mitochondrial Biology Unit, UK John E. Walker**, MRC Mitochondrial Biology Unit, UK Ian Holt, MRC National Institute for Medical Research, UK Douglas C. Wallace**, Children's Hospital of Philadelphia & Howy Jacobs, University of Helsinki, Finland University of Pennsylvania Laurie Kaguni, Michigan State University Mila Pollock**, Cold Spring Harbor Laboratory Emine Koç, Marshall University Since it was first observed within cells at the end of the nineteenth century, our bacterial endosymbiont, the Carla Koehler, University of California, Los Angeles mitochondrion, has been interrogated from many perspectives. Initially described as a cytoplasmic ** Also Session Chairs & Speakers Edmund Kunji, MRC Mitochondrial Biology Unit, UK structure, then as the source of energy, later an organismal entity, and recently a component of many Nick Lane, University College London, UK diseases, the multi-faceted mitochondrion has engendered fascination from a broad spectrum of physical, Session Chairs Nils-Göran Larsson, Karolinska Institute, Sweden chemical, biological, and medical perspectives. Jennifer Lippincott-Schwartz, -

Annual Report 2014 Years
Annual Report 2014 years IST Austria Annual Report 2014 Content 5 years of networking As IST Austria celebrated its fifth anniversary in 2014, this annual report not just reviews the last year but also provides facts, figures, and numbers relevant to the first five years of IST Austria. forewords Scientists at IST Austria rely on professional networks to conduct 02 Foreword by the President excellent research. They work in teams and collaborate with research 03 Guest Commentary by Arnold Schmidt groups from numerous universities and institutes in Austria, Europe, and elsewhere. They meet colleagues at international events to exchange the institute ideas and share their findings. As academic networking is crucial for 04 IST Austria at a Glance scientists at IST Austria, “network” has been chosen as an ancillary theme for the 2014 annual report. 5 years IST Austria 08 Interview Nick Barton: The 1st to Accept the Challenge 10 Recognizing 5 Years of Excellence 11 Science is a Network 12 PhD Students and Postdocs at IST Austria research 14 Research Highlight Evolutionary Biology 16 Current Research at IST Austria 18 Barton Group 19 Benková Group 20 Bollback Group 21 Bollenbach Group 22 Chatterjee Group 23 Cremer Group 24 Csicsvari Group 25 Edelsbrunner Group 26 Erdős Group 27 Friml Group 28 Guet Group 29 Heisenberg Group 30 Henzinger Group 31 Hippenmeyer Group 32 Hof Group 33 Janovjak Group 34 Jonas Group 35 Kolmogorov Group 36 Lampert Group 37 Lemeshko Group 38 Maas Group 39 Novarino Group 40 Pietrzak Group 41 Seiringer Group 42 Shigemoto -
EMBO Members Meeting 2019 Programme Tuesday 29 OCTOBER
29 –31 OCTOBER 2019 EMBO Members Meeting 2019 Programme Tuesday 29 OCTOBER 08:00 - 09:00 REGISTRATION 09:00 - 09:10 Maria Leptin Welcome address EMBO DIRECTOR SESSION 1 Howard Riezman University of Geneva, CH CHAIR 09:10 Blanche Schwappach Physiological integration of the COPI vesicle coat University Medical Center Göttingen, DE 09:30 Aurélien Roux Common principles of surface remodelling in cell membrane traffic and epithelium folding University of Geneva, CH Proposer: Howard Riezman 09:50 Robert Tampé How supramolecular chaperone and translocation complexes shape our adaptive immunity Goethe University Frankfurt, DE 10:10 Katrin Rittinger Ubiquitin-dependent control of cell signalling Francis Crick Institute, Cambridge, GB 10:30 Frank Jülicher Biophysics of active systems Max Planck Institute for the Physics of Complex Systems, Dresden, DE Proposer: Stephan Grill 10:50 - 11:10 COFFEE BREAK: EMBL OPERON FOYER SESSION 2 Carsten Janke Institut Curie, Orsay, FR CHAIR 11:10 Nynke H. Dekker Studies of replication at the single-molecule level TU Delft, NL 11:30 Andrzej Dziembowski Regulation of Gene Expression by non-canonical poly(A) and poly(U) polymerases Polish Academy of Sciences, Warsaw, PL 11:50 Tadatsugu Taniguchi Dead cell-derived molecules in inflammation and cancer University of Tokyo, JP 12:10 Paul Lehner The HUSH epigenetic transcriptional complex — defending the genome from retroelement attack University of Cambridge, GB 12:30 - 13:30 LUNCH: EMBL OPERON FOYER SESSION 3 Duncan Odom DKFZ, Heidelberg, DE CHAIR 13:30 Adèle Marston Mechanisms of chromosome segregation University of Edinburgh, GB 13:50 James Turner Sex chromosomes in development and disease Francis Crick Institute, Cambridge, GB 14:10 Fanni Gergely Precision, speed and size – how centrosomes fine-tune cell division and development University of Cambridge, GB Proposer: Duncan Odom 14:30 G. -
Book Title Author Copyright Year Editore N.1 (Endo)Symbiotic Methanogenic Archaea Johannes H.P
SBBL - SPRINGER E-BOOKS : TITOLI CORRENTI 2013 Book Title Author Copyright Year Editore n.1 (Endo)symbiotic Methanogenic Archaea Johannes H.P. Hackstein 2010 Springer n.2 ‘In Considerable Variety’: Introducing the Diversity of Australia’s Insects Tim R. New 2011 Springer n.3 25 Years of p53 Research Pierre Hainaut, Klas G. Wiman 2005 Springer n.4 34th Hemophilia Symposium I. Scharrer, W. Schramm 2005 Springer n.5 35th Hemophilia Symposium Inge Scharrer, Wolfgang Schramm 2006 Springer n.6 36th Hemophilia Symposium Hamburg 2005 Inge Scharrer, Wolfgang Schramm 2007 Springer n.7 37th Hemophilia Symposium Inge Scharrer, Wolfgang Schramm 2008 Springer n.8 41es Journées nationales de la Société Française de Médecine Périnatale (Grenoble 12–14 octobre 2011) Société Française de Médecine Périnatale 2011 Springer n.9 42es Journées nationales de la Société Française de Médecine Périnatale (Montpellier 17–19 octobre 2012) Société Française de Médecine Périnatale 2013 Springer n.10 55 Years German Society of Anaesthesiology and Intensive Care Medicine J. Schüttler 2012 Springer n.11 5-HT2C Receptors in the Pathophysiology of CNS Disease Giuseppe Di Giovanni, Ennio Esposito, Vincenzo Di Matteo 2011 Springer n.12 60 Years of Survival Outcomes at The University of Texas MD Anderson Cancer Center M. Alma Rodriguez, Ronald S. Walters, Thomas W. Burke 2013 Springer n.13 7.0 Tesla MRI Brain Atlas Zang-Hee Cho 2010 Springer n.14 99mTc-Sestamibi Jan Bucerius, Hojjat Ahmadzadehfar, Hans-Jürgen Biersack 2012 Springer n.15 A Blueprint for Promoting Academic and Social Competence in After-School Programs Thomas P. Gullotta, Martin Bloom, Christianne F. -
Structure and Mechanism of the Mrp Complex, an Ancient Cation/Proton Antiporter Julia Steiner, Leonid Sazanov*
RESEARCH ARTICLE Structure and mechanism of the Mrp complex, an ancient cation/proton antiporter Julia Steiner, Leonid Sazanov* Institute of Science and Technology Austria, Klosterneuburg, Austria Abstract Multiple resistance and pH adaptation (Mrp) antiporters are multi-subunit Na+ (or K+)/ H+ exchangers representing an ancestor of many essential redox-driven proton pumps, such as respiratory complex I. The mechanism of coupling between ion or electron transfer and proton translocation in this large protein family is unknown. Here, we present the structure of the Mrp complex from Anoxybacillus flavithermus solved by cryo-EM at 3.0 A˚ resolution. It is a dimer of seven-subunit protomers with 50 trans-membrane helices each. Surface charge distribution within each monomer is remarkably asymmetric, revealing probable proton and sodium translocation pathways. On the basis of the structure we propose a mechanism where the coupling between sodium and proton translocation is facilitated by a series of electrostatic interactions between a cation and key charged residues. This mechanism is likely to be applicable to the entire family of redox proton pumps, where electron transfer to substrates replaces cation movements. Introduction The Na+/H+ antiporters are widely distributed secondary active transporters that use the proton motive force to efflux intracellular sodium ions (Ito et al., 2017). Several protein families catalyse *For correspondence: this reaction and are mostly encoded by a single gene, such as the NHE family in eukaryotes and the [email protected] NhaA family in bacteria (Krulwich et al., 2011). Mrp antiporters are unique as they usually consist of seven subunits (MrpABCDEFG) encoded in a single operon (Figure 1—figure supplement 1a). -
![Rigaku Americas E-Mail: Rinttyo@Rigaku.Co.Jp E-Mail: Info@Rigaku.Com Tel: +[81] 3-3479-0618 Tel: (281) 362-2300 FAX: +[81] 3-3479-6112 FAX: (281) 364-3628](https://docslib.b-cdn.net/cover/6806/rigaku-americas-e-mail-rinttyo-rigaku-co-jp-e-mail-info-rigaku-com-tel-81-3-3479-0618-tel-281-362-2300-fax-81-3-3479-6112-fax-281-364-3628-7686806.webp)
Rigaku Americas E-Mail: [email protected] E-Mail: [email protected] Tel: +[81] 3-3479-0618 Tel: (281) 362-2300 FAX: +[81] 3-3479-6112 FAX: (281) 364-3628
Protein Crystallography Newsletter Volume 3, No. 8, August 2011 Crystallography in the news In this issue: August 2, 2011. Dr. Kalle Gehring, a professor in McGill University's Department of Crystallography in the news Biochemistry, has been awarded a Pilot Project Grant by the Parkinson Society of Photo of the month Canada (PSC) for his ongoing work that looks at the molecular and cellular processes that underlie neurological diseases like Parkinson's Disease (PD). Call for nominations: Carl Brändén Award BioSAXS-1000 Kratky camera August 03, 2011 Mitegen LLC, a provider of innovative consumables for X-ray diffraction, crystallography and protein crystallization to academic, pharmaceutical, industrial and Spotlight: UTMB, Sealy Center government researchers around the world, announced that it has signed an agreement Useful links for crystallography with AP Innovations to distribute their first product, the Quick Puck Loader for the Professional education opportunities Rigaku ACTOR robot. Science video of the month August 8, 2011. Coupling laser-driven, two-dimensional fluorescence imaging and high- Funny video of the month performance computer modeling, a six-member team - led by University of Oregon chemist Andrew H. Marcus and Harvard University chemist Alan Aspuru-Guzik - solved Last month's survey results the conformation of self-assembled porphyrin molecules in a biological membrane. Survey question of the month August crystallographic papers August 10, 2011. The availability of a postdoctoral fellowship was announced for the laboratory of Dr Ian Taylor (MRC National Institute for Medical Research, London), to Book reviews study the structure and mechanism of the recently described retroviral restriction factor SAMHD1. The primary focus of the project will be to establish the relationship between the biochemical and enzymatic properties of SAMHD1 and the mechanism of virus Mark Your Calender restriction. -
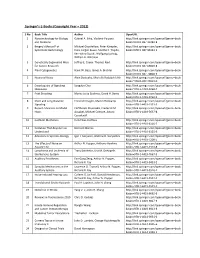
Springer's E-Books (Copyright Year – 2012)
Springer’s E‐Books (Copyright Year – 2012) S No Book Title Author OpenURL 1 Nanotechnology for Biology Gabriel A. Silva, Vladimir Parpura http://link.springer.com/openurl?genre=book and Medicine &isbn=978‐0‐387‐31282‐8 2 Bergey’s Manual® of Michael Goodfellow, Peter Kämpfer, http://link.springer.com/openurl?genre=book Systematic Bacteriology Hans‐Jürgen Busse, Martha E. Trujillo, &isbn=978‐0‐387‐95043‐3 Ken‐ichiro Suzuki, Wolfgang Ludwig, William B. Whitman 3 Genetically Engineered Mice Jeffrey E. Green, Thomas Ried http://link.springer.com/openurl?genre=book for Cancer Research &isbn=978‐0‐387‐69803‐8 4 Plant Cytogenetics Hank W. Bass, James A. Birchler http://link.springer.com/openurl?genre=book &isbn=978‐0‐387‐70868‐3 5 Neuronal Noise Alain Destexhe, Michelle Rudolph‐Lilith http://link.springer.com/openurl?genre=book &isbn=978‐0‐387‐79019‐0 6 Encyclopedia of Signaling Sangdun Choi http://link.springer.com/openurl?genre=book Molecules &isbn=978‐1‐4419‐0460‐7 7 Fruit Breeding Marisa Luisa Badenes, David H. Byrne http://link.springer.com/openurl?genre=book &isbn=978‐1‐4419‐0762‐2 8 Short and Long Distance Friedrich Kragler, Martin Hülskamp http://link.springer.com/openurl?genre=book Signaling &isbn=978‐1‐4419‐1531‐3 9 Recent Advances on Model Eleftherios Mylonakis, Frederick M. http://link.springer.com/openurl?genre=book Hosts Ausubel, Michael Gilmore, Arturo &isbn=978‐1‐4419‐5637‐8 Casadevall 10 Cochlear Mechanics Hendrikus Duifhuis http://link.springer.com/openurl?genre=book &isbn=978‐1‐4419‐6116‐7 11 Evolution That Anyone Can Bernard Marcus http://link.springer.com/openurl?genre=book Understand &isbn=978‐1‐4419‐6125‐9 12 Advances in Systems Biology Igor I.