University of California, San Diego
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CO2, Hothouse and Snowball Earth
CO2, Hothouse and Snowball Earth Gareth E. Roberts Department of Mathematics and Computer Science College of the Holy Cross Worcester, MA, USA Mathematical Models MATH 303 Fall 2018 November 12 and 14, 2018 Roberts (Holy Cross) CO2, Hothouse and Snowball Earth Mathematical Models 1 / 42 Lecture Outline The Greenhouse Effect The Keeling Curve and the Earth’s climate history Consequences of Global Warming The long- and short-term carbon cycles and silicate weathering The Snowball Earth hypothesis Roberts (Holy Cross) CO2, Hothouse and Snowball Earth Mathematical Models 2 / 42 Chapter 1 Historical Overview of Climate Change Science Frequently Asked Question 1.3 What is the Greenhouse Effect? The Sun powers Earth’s climate, radiating energy at very short Earth’s natural greenhouse effect makes life as we know it pos- wavelengths, predominately in the visible or near-visible (e.g., ul- sible. However, human activities, primarily the burning of fossil traviolet) part of the spectrum. Roughly one-third of the solar fuels and clearing of forests, have greatly intensifi ed the natural energy that reaches the top of Earth’s atmosphere is refl ected di- greenhouse effect, causing global warming. rectly back to space. The remaining two-thirds is absorbed by the The two most abundant gases in the atmosphere, nitrogen surface and, to a lesser extent, by the atmosphere. To balance the (comprising 78% of the dry atmosphere) and oxygen (comprising absorbed incoming energy, the Earth must, on average, radiate the 21%), exert almost no greenhouse effect. Instead, the greenhouse same amount of energy back to space. Because the Earth is much effect comes from molecules that are more complex and much less colder than the Sun, it radiates at much longer wavelengths, pri- common. -

Keeling Curve: Result, Interpretation & Global Monitoring
International Journal for Empirical Education and Research Keeling Curve: Result, Interpretation & Global Monitoring Augustyn Ostrowski Faculty of Geography & Geology Jagiellonian University Email: [email protected] (Author of Correspondence) Poland Abstract The Keeling Curve is a graph of the accumulation of carbon dioxide in the Earth's atmosphere based on continuous measurements taken at the Mauna Loa Observatory on the island of Hawaii from 1958 to the present day. The curve is named for the scientist Charles David Keeling, who started the monitoring program and supervised it until his death in 2005. Keywords: Mauna Loa Measurements; Results and Interpretation; Global Monitoring; Ralph Keeling. ISSN Online: 2616-4833 ISSN Print: 2616-4817 35 1. Introduction Keeling's measurements showed the first significant evidence of rapidly increasing carbon dioxide levels in the atmosphere. According to Dr Naomi Oreskes, Professor of History of Science at Harvard University, the Keeling curve is one of the most important scientific works of the 20th century. Many scientists credit the Keeling curve with first bringing the world's attention to the current increase of carbon dioxide in the atmosphere. Prior to the 1950s, measurements of atmospheric carbon dioxide concentrations had been taken on an ad hoc basis at a variety of locations. In 1938, engineer and amateur meteorologist Guy Stewart Callendar compared datasets of atmospheric carbon dioxide from Kew in 1898-1901, which averaged 274 parts per million by volume (ppm), and from the eastern United States in 1936-1938, which averaged 310 ppmv, and concluded that carbon dioxide concentrations were rising due to anthropogenic emissions. However, Callendar's findings were not widely accepted by the scientific community due to the patchy nature of the measurements. -

This Is Nature; This Is Un-Nature: Reading the Keeling Curve
Joshua P. Howe Downloaded from https://academic.oup.com/envhis/article-abstract/20/2/286/528915 by OUP site access user on 24 May 2019 This Is Nature; This Is Un-Nature: Reading the Keeling Curve Data images make odd cultural artifacts. On one hand, scientists pre- sent their data in images as a form of visual communication, intended, like other forms of visual culture, to convey both specific information and larger culturally coded messages. On the other hand, scientists typically hew to methods of measurement and math- ematical analysis intended to ensure that the data they present reflect some objective reality that transcends the cultural. To the extent that the data tell a cultural story, it is supposed to “speak for itself.” Such is the case with the Keeling Curve, the oscillating upward- sloping graph of measured atmospheric carbon dioxide (CO2) that has come to stand as one of the most important and powerful scien- tific symbols of anthropogenic climate change. To a lay reader, it may seem odd to read a simple measure of atmospheric gas through the many-sided prism of modern American life the way you might read a historical photograph or piece of art. And yet the Keeling Curve func- tions as much as a symbol in our collective cultural understanding of climate change as it does a representation of data about CO2. The Keeling Curve faithfully represents something quite real—the accumulation of CO2 in the atmosphere since 1958, expressed in parts per million (ppm)—but it is also a constructed image ripe for reading, similar to a painting, a photograph, a landscape, or a written document. -

Biogeochemical Cycles and Our Environment
Name: ________________________________________ TOC#_____ Biogeochemical Cycles and Our Environment Water, Carbon, and Nitrogen cycle throughout our plant. The way in which this occurs, and at what rate will play a large role in our environment and our ecosystem. If one part of the cycle changes (either increases or decreases) that will create a shift in the cycle. 1. Look at page 96 in your text book. Draw the water cycle (you only need words and arrows-not drawings of trees etc). 2. If the rate of evaporation increased, what would be the change to the environment? 3. Look at page 98 in your text book. Draw the carbon cycle (you only need words and arrows-not drawings of trees etc). 4. If the rate of burning fossil fuels/human activity increased, what would be the change to the environment? 5. Look at page 99 in your text book. Draw the nitrogen cycle (you only need words and arrows-not drawings of trees etc). 6. If there was more nitrogen in the atmosphere, how would the cycle change? 7. Read the article below (please note the date of the article!). As you read, please underline anything you find interesting. If something is confusing, but a ? next to that paragraph. May 10, 2013 (New York Times) Heat-Trapping Gas Passes Milestone, Raising Fears By JUSTIN GILLIS The level of the most important heat-trapping gas in the atmosphere, carbon dioxide, has passed a long-feared milestone, scientists reported Friday, reaching a concentration not seen on the earth for millions of years. Scientific instruments showed that the gas had reached an average daily level above 400 parts per million — just an odometer moment in one sense, but also a sobering reminder that decades of efforts to bring human- produced emissions under control are faltering. -

A Climate Chronology Sharon S
Landscape of Change by Jill Pelto A Climate Chronology Sharon S. Tisher, J.D. School of Economics and Honors College University of Maine http://umaine.edu/soe/faculty-and-staff/tisher Copyright © 2021 All Rights Reserved Sharon S. Tisher Foreword to A Climate Chronology Dr. Sean Birkel, Research Assistant Professor & Maine State Climatologist Climate Change Institute School of Earth and Climate Sciences University of Maine March 12, 2021 The Industrial Revolution brought unprecedented innovation, manufacturing efficiency, and human progress, ultimately shaping the energy-intensive technological world that we live in today. But for all its merits, this transformation of human economies also set the stage for looming multi-generational environmental challenges associated with pollution, energy production from fossil fuels, and the development of nuclear weapons – all on a previously unimaginable global scale. More than a century of painstaking scientific research has shown that Earth’s atmosphere and oceans are warming as a result of human activity, primarily through the combustion of fossil fuels (e.g., oil, coal, and natural gas) with the attendant atmospheric emissions of carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), and other * greenhouse gases. Emissions of co-pollutants, such as nitrogen oxides (NOx), toxic metals, and volatile organic compounds, also degrade air quality and cause adverse human health impacts. Warming from greenhouse-gas emissions is amplified through feedbacks associated with water vapor, snow and sea-ice -

Scripps Carbon Dioxide and Oxygen Programs Ralph F
Scripps Carbon Dioxide and Oxygen Programs Ralph F. Keeling, PI, Scripps Institution of Oceanography, UCSD, La Jolla, CA Scripps CO2 Program - www.scrippsco2.ucsd.edu Scripps O2 Program - http://bluemoon.ucsd.edu Ralph Keeling - [email protected] CO TREND O TREND CO EXTRACTION RACK INTRODUCTION 4 2 5 2 9 2 89 90 91 92 93 94 95 96 97 98 99 00 01 02 03 04 05 06 0 0 System for extracting CO2 -50 -50 from flasks into glass am- ALT -100 -100 poules. CO is trapped cryo- The Scripps CO2 program was initiated in 1956 by Charles David Keeling and 2 82N operated under his direction until his passing in 2005. It is currently being con- -150 -150 genically from the air using -200 -200 liquid nitrogen, and then tinued by Ralph F. Keeling, who also runs the parallel Scripps O2 program. The -250 transferred into a 1/4" O.D. combined programs are sustaining the longest continuous measurements of -300 glass tube. The tube is then changes in atmospheric carbon dioxide concentration, the isotopes of CO , and -100 -350 sealed using an electronic 2 CBA -150 -400 heating element. Examples of 55N -200 the atmospheric O2 concentration (as O2/N2 ratio). The program involves flask -450 the glass ampoules are shown collections made through cooperative programs with field stations at roughly a 0 -250 in the insert. The system -50 -300 allows for up to six flask to be dozen stations around the world. The emphasis in the program is in the detec- LJO -100 -350 33 N extracted in series. -

A US Carbon Cycle Science Plan
A U.S. CARBON CYCLE SCIENCE PLAN A Report of thethe Carbon and Climate Working Group Jorge L. Sarmiento and Steven C. Wofsy, Co-Chairs A U.S. CARBON CYCLE SCIENCE PLAN A Report of the Carbon and Climate Working Group Jorge L. Sarmiento and Steven C.Wofsy, Co-Chairs Prepared at the Request of the Agencies of the U. S. Global Change Research Program 1999 The Carbon Cycle Science Plan was written by the Carbon and Climate Working Group consisting of: Jorge L. Sarmiento,Co-Chair Princeton University Steven C.Wofsy, Co-Chair Harvard University Eileen Shea,Coordinator Adjunct Fellow, East-West Center A.Scott Denning Colorado State University William Easterling Pennsylvania State University Chris Field Carnegie Institution Inez Fung University of California,Berkeley Ralph Keeling Scripps Institution of Oceanography, University of California, San Diego James McCarthy Harvard University Stephen Pacala Princeton University W. M. Post Oak Ridge National Laboratory David Schimel National Center for Atmospheric Research Eric Sundquist United States Geological Survey Pieter Tans NOAA,Climate Monitoring and Diagnostics Laboratory Ray Weiss Scripps Institution of Oceanography, University of California,San Diego James Yoder University of Rhode Island Preparation of the report was sponsored by the following agencies of the United States Global Change Research Program: the Department of Energy, the National Aeronautics and Space Administration,the National Oceanic and Atmospheric Administration,the National Science Foundation,and the United States Geological Survey of the Department of Interior. The Working Group began its work in early 1998,holding meetings in March and May 1998. A Carbon Cycle Science Workshop was held in August 1998, in Westminster, Colorado,to solicit input from the scientific com- munity, and from interested Federal agencies. -

Textor 19 (France, Germany), Ray Wang (USA), Ray Weiss (USA), Tim Whorf (USA) 20 21 Review Editors: Teruyuki Nakajima (Japan), V
Final Draft Chapter 2 IPCC WG1 Fourth Assessment Report 1 Chapter 2: Changes in Atmospheric Constituents and in Radiative Forcing 2 3 Coordinating Lead Authors: Piers Forster (UK), V. Ramaswamy (USA) 4 5 Lead Authors: Paulo Artaxo (Brasil), Terje Berntsen (Norway), Richard A. Betts (UK), David W. Fahey 6 (USA), James Haywood (UK), Judith Lean (USA), David C. Lowe (New Zealand), Gunnar Myhre 7 (Norway), John Nganga (Kenya), Ronald Prinn (USA, New Zealand), Graciela Raga (Mexico, Argentina), 8 Michael Schulz (France, Germany), Robert Van Dorland (Netherlands) 9 10 Contributing Authors: Greg Bodeker (New Zealand), Olivier Boucher (UK, France), William Collins 11 (USA), Thomas J. Conway (USA), Ed Dlugokencky (USA), James W. Elkins (USA), David Etheridge 12 (Australia), Peter Foukal (USA), Paul Fraser (Australia), Marvin Geller (USA), Fortunat Joos (Switzerland), 13 C. David Keeling (USA), Ralph Keeling (USA), Stefan Kinne (Germany), Keith Lassey (New Zealand), 14 Ulrike Lohmann (Switzerland), Andrew Manning (UK, New Zealand), Steve Montzka (USA), David Oram 15 (UK), Kath O'Shaughnessy (New Zealand), Stephen Piper (USA), Gian-Kasper Plattner (Switzerland), 16 Michael Ponater (Germany), Navin Ramankutty (USA, India), George Reid (USA), David Rind (USA), 17 Karen Rosenlof (USA), Robert Sausen (Germany), Dan Schwarzkopf (USA), S. Solanki (Germany), 18 Georgiy Stenchikov (USA), Nicola Stuber (UK, Germany), Toshihiko Takemura (Japan), Christiane Textor 19 (France, Germany), Ray Wang (USA), Ray Weiss (USA), Tim Whorf (USA) 20 21 Review Editors: -

Simplified Carbon Cycle
Carbon-cycle Biogeochemistry SWES 410/510 March 7, 2014 I. Simplified C-cycle II. Carbon dioxide III.Methane Simplified carbon cycle Phototrophic fixation CO2 ‐ reduction (+4) Respiration O2 Methanotrophic ‐ oxidation CH2O ‐ oxidation organic (0) Respiration & fermentation Acetoclastic & ‐ oxidation methylotrophic methanogenesis ‐ reduction CO2 (+4) CH 2H Methanotrophic 4 ‐ oxidation (‐4) CO2 reduction methanogenesis anO Source: R. Mondav 2 1 A pressing question What is the fate of all that fossil fuel CO2? Atmosphere? Ocean? Charles David Keeling. Above: 1961 Right: in 1990s A pressing question What is the fate of all that fossil fuel CO2? Atmosphere? Ocean? Charles David Keeling. Made the first high-accuracy measurements of atmospheric CO2 in a sufficiently remote place (the south pole) to be minimally influenced by transients or local biases. Above: 1961 Right: in 1990s 2 A pressing question What is the fate of all that fossil fuel CO2? Atmosphere? Ocean? Charles David Keeling. Made the first high-accuracy measurements of atmospheric CO2 in a sufficiently remote place (the south pole) to be minimally influenced by transients or local biases. A rising level of CO2 in the atmosphere was first demonstrated in 1960 in Above: 1961 Antarctica, visible after only two years of Right: in 1990s measurements. (Keeling, 1960) 3 Present: where does all the carbon go? 4 Uptake by = land and oceans Questions about carbon uptake Part II. Where does all the carbon go? 1. How do we tell how much is going into the land, and how much is going into the ocean? 2. What causes the high interannual variability in atm. CO2? (the wiggles?) Part III. -
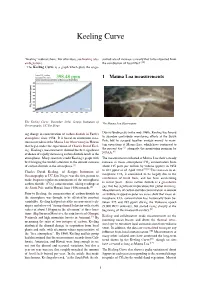
Keeling Curve
Keeling Curve “Keeling” redirects here. For other uses, see Keeling (dis- served rate of increase is nearly that to be expected from ambiguation). the combustion of fossil fuel”.[4] The Keeling Curve is a graph which plots the ongo- 1 Mauna Loa measurements The Keeling Curve, December 2014, Scripps Institution of The Mauna Loa Observatory Oceanography, UC San Diego ing change in concentration of carbon dioxide in Earth’s Due to funding cuts in the mid-1960s, Keeling was forced atmosphere since 1958. It is based on continuous mea- to abandon continuous monitoring efforts at the South surements taken at the Mauna Loa Observatory in Hawaii Pole, but he scraped together enough money to main- that began under the supervision of Charles David Keel- tain operations at Mauna Loa, which have continued to the present day [5] alongside the monitoring program by ing. Keeling’s measurements showed the first significant [6] evidence of rapidly increasing carbon dioxide levels in the NOAA. atmosphere. Many scientists credit Keeling’s graph with The measurements collected at Mauna Loa show a steady first bringing the world’s attention to the current increase increase in mean atmospheric CO2 concentration from of carbon dioxide in the atmosphere.[1] about 315 parts per million by volume (ppmv) in 1958 [7][8] Charles David Keeling, of Scripps Institution of to 401 ppmv as of April 2014. This increase in at- mospheric CO is considered to be largely due to the Oceanography at UC San Diego, was the first person to 2 make frequent regular measurements of the atmospheric combustion of fossil fuels, and has been accelerating in recent years. -

Charles David Keeling Papers
http://oac.cdlib.org/findaid/ark:/13030/c83j3ktw No online items Charles David Keeling Papers Special Collections & Archives, UC San Diego Special Collections & Archives, UC San Diego Copyright 2019 9500 Gilman Drive La Jolla 92093-0175 [email protected] URL: http://libraries.ucsd.edu/collections/sca/index.html Charles David Keeling Papers SMC 0099 1 Descriptive Summary Languages: English Contributing Institution: Special Collections & Archives, UC San Diego 9500 Gilman Drive La Jolla 92093-0175 Title: Charles David Keeling Papers Creator: Keeling, Charles D., 1928-2005 Identifier/Call Number: SMC 0099 Physical Description: 117.6 Linear feet(218 archives boxes, 2 card file boxes, 28 records cartons, 2 flat boxes, and 4 map case folders) Physical Description: 1 GBof digital files Date (inclusive): 1934-2006 (bulk 1955-2000) Abstract: Papers of Charles David Keeling (1928-2005), an environmental chemist, founder of the Scripps CO₂ Program, and professor emeritus at UC San Diego. Dr. Keeling was considered the world's leading authority on atmospheric greenhouse gases and climate science. The collection includes correspondence, writings and reports, teaching and lecture materials, and documentation and data from the Scripps CO₂ Program. Scope and Content of Collection Papers of Charles David Keeling (1928-2005), an environmental chemist, founder of the Scripps CO₂ Program, and professor emeritus at UC San Diego. Dr. Keeling was considered the world's leading authority on atmospheric greenhouse gases and climate science. The collection documents decades of Keeling's meticulous measurement of atmospheric CO₂ at land-based stations and on shipboard expeditions, his founding of and continuous leadership for the Scripps CO₂ Program, and his research. -

Chapter 2 Changes in Atmospheric Constituents and Radiaitve Forcing
Second-Order Draft Chapter 2 IPCC WG1 Fourth Assessment Report 1 Chapter 2: Changes in Atmospheric Constituents and in Radiative Forcing 2 3 Coordinating Lead Authors: Piers Forster (UK), V. Ramaswamy (USA) 4 5 Lead Authors: Paulo Artaxo, Terje Berntsen, Richard Betts, Dave Fahey, Jim Haywood, Judith Lean, Dave 6 Lowe, Gunnar Myhre, John Nganga, Ronald Prinn, Graciela Raga, Michael Schulz, Rob Van Dorland 7 8 Contributing Authors: Greg Bodeker (New Zealand), Olivier Boucher (France), William Collins (USA), 9 Thomas J. Conway (USA), Ed Dlugokencky (USA), Jim Elkins (USA), David Etheridge (Australia), Paul 10 Fraser (Australia), Dave Keeling (USA),, Ralph Keeling (USA), Stefan Kinne (Germany), Keith Lassey 11 (New Zealand), Ulrike Lohmann (Switzerland), Andrew Manning (UK), Steve Montzka (USA), David Oram 12 (UK), Kath O'Shaughnessy (New Zealand), Steve Piper (USA), Michael Ponater (Germany), Navin 13 Ramankutty (India), Karen Rosenlof (USA), Robert Sausen (Germany), Dan Schwarzkopf (USA), Georgiy 14 Stenchikov (USA), Nicola Stuber (Germany), Toshihiko Takemura (Japan). Christiane Textor (France), Ray 15 Wang (USA), Ray Weiss (USA), Tim Whorf (USA). 16 17 Review Editors: Teruyuki Nakajima (Japan), V. Ramanathan (USA) 18 19 Date of Draft: 28 February 2006 20 21 Notes: TSU compiled version 22 23 24 Table of Contents 25 26 Executive Summary............................................................................................................................................................3 27 2.1 Introduction and Scope ..............................................................................................................................................8