Gene Expression Analysis Reveals Novel Gene Signatures Between Young and Old Adults in Human Prefrontal Cortex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cytotoxicity and Genome-Wide Microarray Analysis of Intestinal Smooth Muscle Cells in Response to Hexavalent Chromium Induction
Zoological Research 34 (E3): E93−E100 DOI:10.11813/j.issn.0254-5853.2013.E3.E93 Cytotoxicity and genome-wide microarray analysis of intestinal smooth muscle cells in response to hexavalent chromium induction Li-Fang JIN, Yuan-Yuan WANG, Zi-Dong ZHANG, Yi-Meng YUAN, Yi-Rui HU, Yang-Feng WEI, * Jian NI College of Life Science of Shaoxing University, Shaoxing Zhejiang 312000, China Abstract: Chronic ingestion of high concentrations of hexavalent chromium [Cr(VI)] in drinking water induces intestinal tumors in mice; however, information on its toxicity on intestinal smooth muscle cells is limited. The present study aimed to assess the in vitro and in vivo toxicological effects of Cr(VI) on intestinal smooth muscle cells. Human intestinal smooth muscle cells (HISM cells) were cultured with different concentrations of Cr(VI) to evaluate effects on cell proliferation ability, oxidative stress levels, and antioxidant system. Furthermore, tissue sections in Cr(VI) exposed rabbits were analyzed to evaluate toxicity on intestinal muscle cells in vivo. Gene chips were utilized to assess differential gene expression profiles at the genome-wide level in 1 μmol/L Cr(VI) treated cells. Intestinal tissue biopsy results showed that Cr(VI) increased the incidences of diffuse epithelial hyperplasia in intestinal jejunum but caused no obvious damage to the structure of the muscularis. Cell proliferation analysis revealed that high concentrations (≥64 μmol/L) but not low concentrations of Cr(VI) (≤16 μmol/L) significantly inhibited the growth of HISM cells. For oxidative stress levels, the expression of reactive oxygen species (ROS) and nitric oxide (NO) was elevated at high concentrations (≥64 μmol/L) but not at low concentrations of Cr(VI) (≤16 μmol/L). -

Genetics of Distal Hereditary Motor Neuropathies
GENETICSOFDISTALHEREDITARY MOTOR NEUROPATHIES By alexander peter drew A thesis submitted for the Degree of Doctor of Philosophy Supervised by Professor Garth A. Nicholson Dr. Ian P. Blair Faculty of Medicine University of Sydney 2012 statement No part of the work described in this thesis has been submitted in fulfilment of the requirements for any other academic degree or qualification. Except where due acknowledgement has been made, all experimental work was performed by the author. Alexander Peter Drew CONTENTS acknowledgements ............................. i summary .................................... ii list of figures ................................ v list of tables ................................ viii acronyms and abbreviations ..................... xi publications ................................. xiv 1 literature review ........................... 1 1.1 Molecular genetics and mechanisms of disease in Distal Hereditary Motor Neuropathies . .1 1.1.1 Small heat shock protein family . .2 1.1.2 Dynactin 1 (DCTN1).....................9 1.1.3 Immunoglobulin mu binding protein 2 gene (IGHMBP2) 11 1.1.4 Senataxin (SETX)....................... 14 1.1.5 Glycyl-tRNA synthase (GARS)............... 16 1.1.6 Berardinelli-Seip congenital lipodystrophy 2 (SEIPIN) gene (BSCL2)......................... 18 1.1.7 ATPase, Cu2+-transporting, alpha polypeptide gene (ATP7A) 20 1.1.8 Pleckstrin homology domain-containing protein, G5 gene (PLEKHG5)........................... 21 1.1.9 Transient receptor potential cation channel, V4 gene (TRPV4) 22 1.1.10 DYNC1H1 ........................... 23 1.1.11 Clinical variability in dHMN . 24 1.1.12 Common disease mechanisms in dHMN . 29 2 general materials and methods ................. 32 2.1 General materials and reagents . 32 2.1.1 Reagents and Enzymes . 32 2.1.2 Equipment . 33 2.1.3 Plasticware . 33 2.2 Study participants . 34 2.3 DNA methods . -

A Data-Driven Review of the Genetic Factors of Pregnancy Complications
International Journal of Molecular Sciences Article A Data-Driven Review of the Genetic Factors of Pregnancy Complications Yury A. Barbitoff 1,2,3,† , Alexander A. Tsarev 1,4,†, Elena S. Vashukova 3, Evgeniia M. Maksiutenko 2,5, Liudmila V. Kovalenko 6 and Larisa D. Belotserkovtseva 7 and Andrey S. Glotov 3,8,* 1 Bioinformatics Institute, 197342 St. Petersburg, Russia; [email protected] (Y.A.B.); [email protected] (A.A.T.) 2 Department of Genetics and Biotechnology, Saint-Petersburg State University, 199034 St. Petersburg, Russia; [email protected] 3 Department of Genomic Medicine, D.O.Ott Research Institute for Obstetrics, Gynaecology and Reproductology, 199034 St. Petersburg, Russia; [email protected] 4 Department of Biochemistry, Saint-Petersburg State University, 199034 St. Petersburg, Russia 5 St. Petersburg Branch, Vavilov Institute of General Genetics, Russian Academy of Sciences, 199034 St. Petersburg, Russia 6 Department of Pathology, Medical Institute, Surgut State University, 628416 Surgut, Russia; [email protected] 7 Department of Obstetrics, Gynecology and Perinatology, Medical Institute, Surgut State University, 628416 Surgut, Russia; [email protected] 8 Laboratory of Biobanking and Genomic Medicine, Saint-Petersburg State University, 199034 St. Petersburg, Russia * Correspondence: [email protected] † These authors contributed equally to this work. Received: 18 April 2020; Accepted: 6 May 2020; Published: 11 May 2020 Abstract: Over the recent years, many advances have been made in the research of the genetic factors of pregnancy complications. In this work, we use publicly available data repositories, such as the National Human Genome Research Institute GWAS Catalog, HuGE Navigator, and the UK Biobank genetic and phenotypic dataset to gain insights into molecular pathways and individual genes behind a set of pregnancy-related traits, including the most studied ones—preeclampsia, gestational diabetes, preterm birth, and placental abruption. -

Anti-ACTR3B Antibody (ARG57950)
Product datasheet [email protected] ARG57950 Package: 100 μl anti-ACTR3B antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ACTR3B Tested Reactivity Hu Tested Application WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name ACTR3B Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 189-418 of Human ACTR3B (NP_065178.1). Conjugation Un-conjugated Alternate Names ARP3-beta; Actin-related protein ARP4; Actin-related protein 3B; ARP3BETA; Actin-like protein 3B; ARP11 Application Instructions Application table Application Dilution WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control HeLa Calculated Mw 48 kDa Observed Size 47 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol ACTR3B Gene Full Name ARP3 actin-related protein 3 homolog B (yeast) Background This gene encodes a member of the actin-related proteins (ARP), which form multiprotein complexes and share 35-55% amino acid identity with conventional actin. The protein encoded by this gene may have a regulatory role in the actin cytoskeleton and induce cell-shape change and motility. -

Systems Genetics Analysis of the LXS Recombinant Inbred Mouse Strains:Genetic and Molecular Insights Into Acute Ethanol Tolerance
PLOS ONE RESEARCH ARTICLE Systems genetics analysis of the LXS recombinant inbred mouse strains:Genetic and molecular insights into acute ethanol tolerance 1,2 3,4,5 3 3 Richard A. RadcliffeID *, Robin DowellID , Aaron T. Odell , Phillip A. RichmondID , 1 1 6 1 Beth Bennett , Colin LarsonID , Katerina KechrisID , Laura M. SabaID , 7 6 Pratyaydipta RudraID , Shi WenID a1111111111 1 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, CO, United States of America, 2 Institute for Behavioral Genetics, University of Colorado a1111111111 Boulder, Boulder CO, United States of America, 3 BioFrontiers Institute, University of Colorado Boulder, a1111111111 Boulder, CO, United States of America, 4 Department of Molecular, Cellular, and Developmental Biology, a1111111111 University of Colorado Boulder, Boulder, CO, United States of America, 5 Department of Computer Science, a1111111111 University of Colorado Boulder, Boulder, CO, United States of America, 6 Department of Biostatistics and Informatics, Colorado School of Public Health, University of Colorado Anschutz Medical Campus, Aurora, CO, United States of America, 7 Department of Statistics, Oklahoma State University, Stillwater, OK, United States of America * [email protected] OPEN ACCESS Citation: Radcliffe RA, Dowell R, Odell AT, Richmond PA, Bennett B, Larson C, et al. (2020) Abstract Systems genetics analysis of the LXS recombinant inbred mouse strains:Genetic and molecular We have been using the Inbred Long- and Short-Sleep mouse strains (ILS, ISS) and a insights into acute ethanol tolerance. PLoS ONE recombinant inbred panel derived from them, the LXS, to investigate the genetic underpin- 15(10): e0240253. https://doi.org/10.1371/journal. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

A 1.35 Mb DNA Fragment Is Inserted Into the DHMN1 Locus on Chromosome 7Q34–Q36.2
Hum Genet (2016) 135:1269–1278 DOI 10.1007/s00439-016-1720-4 ORIGINAL INVESTIGATION A 1.35 Mb DNA fragment is inserted into the DHMN1 locus on chromosome 7q34–q36.2 Alexander P. Drew1 · Anthony N. Cutrupi1,3 · Megan H. Brewer1,3 · Garth A. Nicholson1,2,3 · Marina L. Kennerson1,2,3 Received: 5 April 2016 / Accepted: 25 July 2016 / Published online: 3 August 2016 © Springer-Verlag Berlin Heidelberg 2016 Abstract Distal hereditary motor neuropathies predomi- for hereditary motor neuropathies and highlights the grow- nantly affect the motor neurons of the peripheral nervous ing importance of interrogating the non-coding genome for system leading to chronic disability. Using whole genome SV mutations in families which have been excluded for sequencing (WGS) we have identified a novel structural genome wide coding mutations. variation (SV) within the distal hereditary motor neuropa- thy locus on chromosome 7q34–q36.2 (DHMN1). The SV involves the insertion of a 1.35 Mb DNA fragment Introduction into the DHMN1 disease locus. The source of the inserted sequence is 2.3 Mb distal to the disease locus at chromo- The distal hereditary motor neuropathies (dHMN) are a some 7q36.3. The insertion involves the duplication of five group of progressive neurodegenerative disorders that pri- genes (LOC389602, RNF32, LMBR1, NOM1, MNX1) and marily affect the motor neurons of distal limbs without partial duplication of UBE3C. The genomic structure of affecting sensory neurons. The disorder is a length depend- genes within the DHMN1 locus are not disrupted by the ant neuropathy in which the longest nerves are initially insertion and no disease causing point mutations within affected. -

Vast Human-Specific Delay in Cortical Ontogenesis Associated With
Supplementary information Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques Supplementary Methods Sample collection We used prefrontal cortex (PFC) and cerebellar cortex (CBC) samples from postmortem brains of 33 human (aged 0-98 years), 14 chimpanzee (aged 0-44 years) and 44 rhesus macaque individuals (aged 0-28 years) (Table S1). Human samples were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, USA, the Netherlands Brain Bank, Amsterdam, Netherlands and the Chinese Brain Bank Center, Wuhan, China. Informed consent for use of human tissues for research was obtained in writing from all donors or their next of kin. All subjects were defined as normal by forensic pathologists at the corresponding brain bank. All subjects suffered sudden death with no prolonged agonal state. Chimpanzee samples were obtained from the Yerkes Primate Center, GA, USA, the Anthropological Institute & Museum of the University of Zürich-Irchel, Switzerland and the Biomedical Primate Research Centre, Netherlands (eight Western chimpanzees, one Central/Eastern and five of unknown origin). Rhesus macaque samples were obtained from the Suzhou Experimental Animal Center, China. All non-human primates used in this study suffered sudden deaths for reasons other than their participation in this study and without any relation to the tissue used. CBC dissections were made from the cerebellar cortex. PFC dissections were made from the frontal part of the superior frontal gyrus. All samples contained an approximately 2:1 grey matter to white matter volume ratio. RNA microarray hybridization RNA isolation, hybridization to microarrays, and data preprocessing were performed as described previously (Khaitovich et al. -
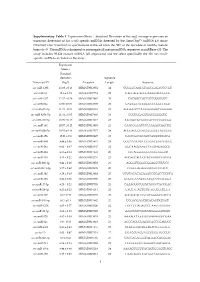
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Detection of H3k4me3 Identifies Neurohiv Signatures, Genomic
viruses Article Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis Liana Basova 1, Alexander Lindsey 1, Anne Marie McGovern 1, Ronald J. Ellis 2 and Maria Cecilia Garibaldi Marcondes 1,* 1 San Diego Biomedical Research Institute, San Diego, CA 92121, USA; [email protected] (L.B.); [email protected] (A.L.); [email protected] (A.M.M.) 2 Departments of Neurosciences and Psychiatry, University of California San Diego, San Diego, CA 92103, USA; [email protected] * Correspondence: [email protected] Abstract: Human postmortem specimens are extremely valuable resources for investigating trans- lational hypotheses. Tissue repositories collect clinically assessed specimens from people with and without HIV, including age, viral load, treatments, substance use patterns and cognitive functions. One challenge is the limited number of specimens suitable for transcriptional studies, mainly due to poor RNA quality resulting from long postmortem intervals. We hypothesized that epigenomic Citation: Basova, L.; Lindsey, A.; signatures would be more stable than RNA for assessing global changes associated with outcomes McGovern, A.M.; Ellis, R.J.; of interest. We found that H3K27Ac or RNA Polymerase (Pol) were not consistently detected by Marcondes, M.C.G. Detection of H3K4me3 Identifies NeuroHIV Chromatin Immunoprecipitation (ChIP), while the enhancer H3K4me3 histone modification was Signatures, Genomic Effects of abundant and stable up to the 72 h postmortem. We tested our ability to use H3K4me3 in human Methamphetamine and Addiction prefrontal cortex from HIV+ individuals meeting criteria for methamphetamine use disorder or not Pathways in Postmortem HIV+ Brain (Meth +/−) which exhibited poor RNA quality and were not suitable for transcriptional profiling. -

1 Major Surgery Induces Acute Changes in DNA
bioRxiv preprint doi: https://doi.org/10.1101/706184; this version posted July 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Major surgery induces acute changes in DNA methylation associated with activation of the immune response. Ryoichi Sadahiro1,2,*, Bridget Knight2,3, Ffion James2, Eilis Hannon2, John Charity3, Ian R. Daniels3, Joe Burrage2, Olivia Knox2, Bethany Crawford2, Neil J. Smart3, Jonathan Mill2* 1 National Cancer Center Japan, Japan 2 University of Exeter Medical School, University of Exeter, United Kingdom 3 Royal Devon & Exeter Hospital, Exeter, United Kingdom * Correspondence to both: - Ryoichi Sadahiro. Address: National Cancer Center Research Institute, National Cancer Center Japan Tsukiji Campus, 5-1-1 Tsukiji, Chuo-ku, Tokyo. 104-0045, Japan. E-mail: [email protected] - Jonathan Mill. Address: University of Exeter Medical School, RILD Building, Royal Devon & Exeter Hospital, Barrack Rd, Exeter. EX2 5DW. UK. E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/706184; this version posted July 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Background: Surgery is an invasive procedure evoking acute inflammatory and immune responses that are believed to mediate risk for postoperative complications including cognitive dysfunction and delirium. -

Next Generation Mapping Reveals Novel Large Genomic Rearrangements in Prostate Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 14), pp: 23588-23602 Research Paper Next generation mapping reveals novel large genomic rearrangements in prostate cancer Weerachai Jaratlerdsiri1,*, Eva K.F. Chan1,2,*, Desiree C. Petersen1,2,*, Claire Yang3, Peter I. Croucher2,4,5, M.S. Riana Bornman6, Palak Sheth3, Vanessa M. Hayes1,2,6,7 1Laboratory for Human Comparative and Prostate Cancer Genomics, Genomics and Epigenetics Division, Garvan Institute of Medical Research, Darlinghurst, Australia 2St Vincent’s Clinical School, University of New South Wales, Randwick, Australia 3Bionano Genomics Inc., San Diego, California, USA 4Bone Biology Division, Garvan Institute of Medical Research, Darlinghurst, Australia 5School of Biotechnology and Biomolecular Sciences, University of New South Wales, Randwick, Australia 6School of Health Systems and Public Health, University of Pretoria, Pretoria, South Africa 7Central Clinical School, University of Sydney, Camperdown, Australia *These authors contributed equally to this work Correspondence to: Vanessa M. Hayes, email: [email protected] Keywords: prostate cancer, structural genomic rearrangements, next generation mapping, next generation sequencing Received: August 15, 2016 Accepted: February 15, 2017 Published: March 01, 2017 ABSTRACT Complex genomic rearrangements are common molecular events driving prostate carcinogenesis. Clinical significance, however, has yet to be fully elucidated. Detecting the full range and subtypes of large structural variants (SVs), greater than