Disseminated Intravascular Coagulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -
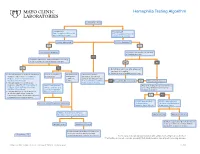
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further. -

Approach to Bleeding Diathesi
Approach to Bleeding Diathesis Dr.Nalini K Pati MD, DNB, DCH (Syd), FRCPA Paediatric Haematologist Royal Children’s Hospital Melbourne Australia Objectives Objectives - I I. Clinical aspects of bleeding Clinical aspects of bleeding II. Hematologic disorders causing bleeding • Coagulation factor disorders • Platelet disorders III. Approach to acquired bleeding disorders • Hemostasis in liver disease • Surgical patients • Warfarin toxicity IV. Approach to laboratory abnormalities • Diagnosis and management of thrombocytopenia V. Drugs and blood products used for bleeding Clinical Features of Bleeding Disorders Petechiae Platelet Coagulation (typical of platelet disorders) disorders factor disorders Site of bleeding Skin Deep in soft tissues Mucous membranes (joints, muscles) (epistaxis, gum, vaginal, GI tract) Petechiae Yes No Ecchymoses (“bruises”) Small, superficial Large, deep Hemarthrosis / muscle bleeding Extremely rare Common Do not blanch with pressure Bleeding after cuts & scratches Yes No (cf. angiomas) Bleeding after surgery or trauma Immediate, Delayed (1-2 days), usually mild often severe Not palpable (cf. vasculitis) Ecchymoses (typical of coagulation factor disorders) Objectives - II Hematologic disorders causing bleeding – Coagulation factor disorders – Platelet disorders Coagulation factor disorders Hemophilia A and B Inherited bleeding Acquired bleeding Hemophilia A Hemophilia B disorders disorders Coagulation factor deficiency Factor VIII Factor IX – Hemophilia A and B – Liver disease – vonWillebrands disease – Vitamin K Inheritance X-linked X-linked recessive recessive – Other factor deficiencies deficiency/warfarin overdose Incidence 1/10,000 males 1/50,000 males –DIC Severity Related to factor level <1% - Severe - spontaneous bleeding 1-5% - Moderate - bleeding with mild injury 5-25% - Mild - bleeding with surgery or trauma Complications Soft tissue bleeding Hemarthrosis (acute) Hemophilia Clinical manifestations (hemophilia A & B are indistinguishable) Hemarthrosis (most common) Fixed joints Soft tissue hematomas (e. -

TPA-2012-2ING:Layout 1
106 Original Article DOI: 10.4274/tpa.823 Glanzmann thrombasthenia: Cerrahpaşa Medical Faculty experience Büşra Kutlubay, Gül Nihal Özdemir, Gülen Tüysüz, Hilmi Apak, Tiraje Celkan İstanbul University Cerrahpaşa Medical Faculty, Department of Pediatrics, Division of Pediatric Hematology-Oncology, İstanbul, Turkey Sum mary Aim: Glazmann thrombasthenia is a rare autosomal recessive disease characterized by a defect in platelet aggregation. Herein, we report the management of children with Glazmann thrombasthenia followed up at Cerrahpasa Medical Faculty Pediatric Hematology Department. Material and Method: The files of nineteen patients (42% girls, 58% boys; median age: 10 months) were retrospectively reviewed. Results: The median age of onset of bleeding symptoms was 9 months (2 weeks-24 months). All patients presented with easy bruising and mucosal bleeding. Fourteen patients’ parents were consanguineous. In 15 patients, flow cytometry was performed. According to this, 7 patients had type I, 6 patients had type II and 2 patients had type III disease. Nine patients were treated with thrombocyte transfusion, tranexamic acid, recombinant active factor VII and fibrin glue as a single or combined therapy; none of them had a major bleeding complication. Conclusions: Bleeding control during invasive procedures may be challenging in children with Glazmann thrombasthenia; local treatments, DDAVP, steroid and antifibrinolytics may be used with success. (Turk Arch Ped 2012; 47: 106-8) Key words: Glanzmann thrombasthenia, thrombocyte function disorder Introduction test, GT platelets do not aggregate following interaction with any substance except for ristocetin. In addition to Glanzmann thrombasthenia (GT) was described by Dr. aggregometer, low than normal levels of glycoprotein (GP) IIb- Eduard Glanzmann in 1918 for the first time as abnormal clot IIIa can be determined with flow cytometer using CD41 and retraction and defects in platelets (1). -

The Patient Is Bleeding & the PT/Aptt Are Normal
The Patient is Bleeding & the PT/aPTT are Normal Dorothy Adcock, MD Title: The patient is bleeding but the APTT and PT are normal As Dr. Worfolk mentioned, the topic of the discussion this morning, the patient is bleeding, but the aPTT and PT are normal. Title: The patient is bleeding but the APTT and PT are normal - Outline What we will discuss this morning is, first of all, we will review the aPTT and PT assays, and I will be referring to the aPTT as just the PTT in the future. We will discuss hemorrhagic disorders of the intrinsic, extrinsic, and common pathway systems that may have a normal PTT and PT. We will then discuss hemorrhagic disorders associated with abnormal fibrin clot formation of the fibrinolytic system. And then finally, we will list disorders of platelets and vasculature that may be associating with bleeding diathesis as well. 1 Title: Screening tests of hemostasis - APTT and PT Now I am certain that all of you are aware that the PTT and PT are the most common screening tests of the hemostatic system. These tests are of course readily available to us in our laboratories; they are familiar to us and inexpensive. And really what I would like to do this morning is bring some more material to us regarding these assays. In the sense that, since these assays have been in the laboratory for such a long period of time, we tend to take these assays for granted, and perhaps expect too much out of these assays. So what I would like to review with you this morning are some of the limitations of these assays. -

Pathophysiology of Bleeding Diathesis in Haemophilia-A
The Egyptian Journal of Medical Human Genetics 19 (2018) 285–295 Contents lists available at ScienceDirect The Egyptian Journal of Medical Human Genetics journal homepage: www.sciencedirect.com Review Pathophysiology of bleeding diathesis in haemophilia-A: A sequential and critical appraisal of non-FVIII related haemostatic dysfunctions and their therapeutic implications ⇑ Umma A. Ibrahim a, Sagir G. Ahmed b, a Department of Paediatrics, Bayero University, PMB 3011, Kano, Kano State, Nigeria b Department of Haematology, Aminu Kano Teaching Hospital, PMB 3452, Kano, Kano State, Nigeria article info abstract Article history: Haemophilia-A is characterized by deficiency of FVIII, but the bleeding diathesis is not a mere reflection Received 29 December 2017 low FVIII activity. The pathophysiology of haemophilic bleeding diathesis is a complex interplay between Accepted 14 January 2018 defective procoagulant function and up-regulated fibrinolysis. Moreover, haemophilic bleeding diathesis Available online 1 February 2018 is frequently compounded by treatment-related and infective complications such as FVIII inhibitors, hep- atitis, HIV infection, non-steroidal anti-inflammatory drugs (NSAID) induced gastritis, and infective Keywords: mucosal injuries such as H pylori gastritis and intestinal and urinary helminthiasis. Hence, pathophysi- Haemophilia ology of haemophilic bleeding is multi-factorial, encompassing both FVIII and non-FVIII haemostatic Bleeding diathesis defects. Currently available literature on pathophysiologic roles of non-FVIII haemostatic defects in hae- Pathophysiology Targeted therapy mophilia is fragmented. This articles is aimed at providing a composite and comprehensive review of the Non-FVIII therapy roles of non-FVIII haemostatic defects and their therapeutic implications in haemophilic bleeding diathe- FVIII by-pass sis, which will enable a holistic approach towards clinical management of the bleeding diathesis. -

Inheritance of Factor VII and Protein S Deficiency Together with Factor V
OLGU SUNUMU / CASE REPORT Kafkas J Med Sci 2016; 6(1):64–68 • doi: 10.5505/kjms.2016.18894 Inheritance of Factor VII and Protein S Deficiency Together with Factor V Leiden Mutation Faktör VII ve Protein S Eksikliğinin Faktör V Leiden Mutasyonu ile Birlikte Kalıtımı Zafer Bıçakcı, Lale Olcay Dr. Abdurrahman Yurtaslan Ankara Oncology Training and Research Hospital, Unit of Pediatric Hematology, Demetevler, Ankara, Turkey ABSTRACT diyatez belirtileri, diğer kalıtsal hemorajik hastalıklarda olduğu gibi Homozygous or heterozygous mutations of factor V Leiden (FV hafifler. Burada, beş ve yedi yaşlarında olup, FVII eksikliği ile bir- Leiden) and the thrombophilic factors like protein S deficiency are likte sırasıyla iki (FV Leiden mutasyonu ve protein S eksikliği) ve associated with venous or arterial thrombosis. In these patients, bir (FV Leiden mutasyonu) trombofilik faktör taşıyan semptomsuz thrombosis may be seen even in the presence of coexistent con- bir kız ve erkek kardeş sunulmaktadır. Faktör VII düzeyleri kız kar- genital disorders of bleeding. Factor VII (FVII) deficiency is a rare deşte % 36 (N: 55–116) ve erkek kardeşte % 38 (N: 52–120) idi. autosomal recessive disorder of blood coagulation. When FVII de- FV Leiden mutasyonu sırasıyla kız ve erkek kardeşte homozigot ve ficiency occurs in combination with thrombophilic mutations, the heterozigottu. Protein S aktivitesi kız kardeşte % 47 (N: 54–118), symptoms of hemorrhagic diathesis are alleviated, like in other in- erkek kardeşte normal idi. Aile çalışmasında, her iki ebeveynde FV herited hemorrhagic disorders. Herein, a 5-year-old and a 7-year- Leiden mutasyonu (heterozigot) ve annede protein S eksikliği [% old, an asymptomatic sister and brother who respectively had 2 51 (N: 55–160)] vardı. -

TEG-Platelet Mapping-Guided Evaluation of Anti-Phospholipid Carrier with Unusual Clinical Presentation and Paradoxical Laboratory Findings
Hematology & Transfusion International Journal Case Report Open Access TEG-platelet mapping-guided evaluation of anti-phospholipid carrier with unusual clinical presentation and paradoxical laboratory findings Abstract Volume 2 Issue 1 - 2016 Anti-phospholipid syndrome is defined by the combination of clinical thrombosis Oksana Volod,1 Yao Ma,1 Sepehr Rokhsar2 and detection of persistent anti-phospholipid antibodies (aPL), including lupus anti- 1Department of Pathology and Laboratory Medicine, Cedars coagulant (LA). Those who do not meet full clinical criteria of overt thrombosis but Sinai Medical Center, USA still harbor circulating aPL can be characterized as “anti-phospholipid carriers.” 2Tower Hematology Oncology Medical Group, USA Regardless of clinical manifestations, aPL can complicate coagulation testing, most commonly by elevating activated partial thromboplastin time (aPTT) and potentially Correspondence: Oksana Volod, Department of Pathology obscuring additional coagulation defects. Platforms for global assays of hemostasis and Laboratory Medicine, Cedars Sinai Medical Center, Los such as thromboelastography (TEG), especially in cases of complex (multiple) Angeles, CA, USA, Tel (310) 423-5471, Fax (310) 423-0483, coagulopathies, grant a wider scope of detection to help guide more specific probing Email [email protected] of individual defects. We report a case of an anti-phospholipid carrier initially evaluated with TEG, revealing paradoxical results suggesting multiple coagulation Received: December 09, 2015 | Published: -

An Approach to the Management of Bleeding Disorders
An approach to the management of bleeding disorders With Dr Shafqat Inam, Haematology Registrar at Concord Hospital and Royal Prince Alfred Hospital Introduction Clotting is dependent on functional platelets, coagulation factors and vasoconstriction. The cause of bleeding diathesis is usually multifactorial in hospital patients with problems with anticoagulants, anti- platelets, congenital disorders and platelet disorders most commonly seen. Patients with bleeding diathesis can present with anaemia due to slow bleeding over time, or a sudden significant bleed, such as a gastrointestinal bleed. When interpreting coagulation studies we need to consider the clinical context. APTT measures therapeutic heparin, PT/INR reflects warfarin therapy; both are for general screens for clotting problems. Platelet count normally is around 150-400 x 10^9/L of blood, but what’s normal also depends in the context. Case 1 - You are a night intern and have been asked to review a patient that has became hypotensive and tachycardic following a right total hip arthroplasty. He is also becoming anxious and pale. There is a large painful haematoma under the surgical site. 1. You are concerned this is an acute post-operative haemorrhage. What would be your approach to this patient? Inspect how the patient looks from the end of the bed and check the observation chart for signs of hypovolemic shock For deteriorating patient, commence resuscitation to stabilise him and conduct rapid investigations in parallel Escalate care to the orthopedic registrar For ongoing bleeding, compress the bleeding site for haemostasis and insert 2 large bore cannulas and commence fluid resuscitation Conduct a history to identify the cause of bleed 2. -

Non-Criteria Manifestations of Juvenile Antiphospholipid Syndrome
Journal of Clinical Medicine Review Non-Criteria Manifestations of Juvenile Antiphospholipid Syndrome Takako Miyamae * and Tomohiro Kawabe Pediatric Rheumatology, Institute of Rheumatology, Tokyo Women’s Medical University, Tokyo 162-8666, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-3-5269-1725 Abstract: Antiphospholipid syndrome (APS) is a systemic autoimmune disorder mainly characterised by increased risks of thrombosis and pregnancy morbidity and persistent positive test results for antiphospholipid antibodies (aPLs). The criteria for diagnosing juvenile APS have yet to be validated, while the Sydney classification criteria do not contain several non-thrombotic clinical manifestations associated with the presence of aPLs. As such, difficulties have been encountered in the diagnosis of patients who have no certain thrombotic occlusions. Moreover, extra-criteria manifestations (i.e., clinical manifestations not listed in the classification criteria), including neurologic manifestations (chorea, myelitis and migraine), haematologic manifestations (thrombocytopenia and haemolytic anaemia), livedo reticularis, nephropathy and valvular heart disease have been reported, which suggests that the clinical spectrum of aPL-related manifestations extends beyond that indicated in the classification criteria. Studies have demonstrated that more than 40% of children with aPLs demonstrated non-thrombotic aPL-related clinical manifestations alone. Moreover, our results showed that the pathogenesis of non-criteria manifestations is characterised by “APS vasculopathy”. The present review introduces the characteristics and findings of non-criteria manifestations observed in juvenile APS. Citation: Miyamae, T.; Kawabe, T. Keywords: antiphospholipid antibodies; children; chorea; systemic lupus erythematosus Non-Criteria Manifestations of Juvenile Antiphospholipid Syndrome. J. Clin. Med. 2021, 10, 1240. https:// doi.org/10.3390/jcm10061240 1.