TEG-Platelet Mapping-Guided Evaluation of Anti-Phospholipid Carrier with Unusual Clinical Presentation and Paradoxical Laboratory Findings
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -
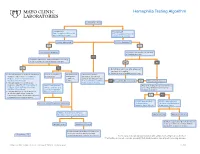
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further. -

ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4
ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4 ISTH 2012 11.6.2012 14:46 Page 1 Table of Contents 3 Welcome Message from the Meeting President 3 Welcome Message from ISTH Council Chairman 4 Welcome Message from SSC Chairman 5 Committees 7 ISTH Future Meetings Calendar 8 Meeting Sponsors 9 Awards and Grants 2012 12 General Information 20 Programme at a Glance 21 Day by Day Scientific Schedule & Programme 22 Detailed Programme Tuesday, 26 June 2012 25 Detailed Programme Wednesday, 27 June 2012 33 Detailed Programme Thursday, 28 June 2012 44 Detailed Programme Friday, 29 June 2012 56 Detailed Programme Saturday, 30 June 2012 68 Hot Topics Schedule 71 ePoster Sessions 97 Sponsor & Exhibitor Profiles 110 Exhibition Floor Plan 111 Congress Centre Floor Plan www.isth.org ISTH 2012 11.6.2012 14:46 Page 2 ISTH 2012 11.6.2012 14:46 Page 3 WelcomeCommittees Messages Message from the ISTH SSC 2012 Message from the ISTH Meeting President Chairman of Council Messages Dear Colleagues and Friends, Dear Colleagues and Friends, We warmly welcome you to the elcome It is my distinct privilege to welcome W Scientific and Standardization Com- you to Liverpool for our 2012 SSC mittee (SSC) meeting of the Inter- meeting. national Society on Thrombosis and Dr. Cheng-Hock Toh and his col- Haemostasis (ISTH) at Liverpool’s leagues have set up a great Pro- UNESCO World Heritage Centre waterfront! gramme aiming at making our off-congress year As setting standards is fundamental to all quality meeting especially attractive for our participants. -

Whole-Exome Sequencing of a Patient with Severe and Complex Hemostatic Abnormalities Reveals a Possible Contributing Frameshift Mutation in C3AR1
Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1 Eva Leinøe1, Ove Juul Nielsen1, Lars Jønson2 and Maria Rossing2∗ Department of Hematology1 and Center for Genomic Medicine2, Rigshospitalet, University of Copenhagen, Blegdamsvej 9, DK-2100 Copenhagen, Denmark Running head: WES reveals a C3AR1 mutation in a complex hemostatic patient ∗Corresponding author: Maria Rossing Center for Genomic Medicine Rigshospitalet University of Copenhagen Blegdamsvej 9 DK-2100 Copenhagen Denmark E-mail: [email protected] Phone: +45 3545 3016 Fax: +45 3545 4435 1 Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The increasing availability of genome-wide analysis has made it possible to rapidly sequence the exome of patients with undiagnosed or unresolved medical conditions. Here, we present the case of a 64-year-old male patient with schistocytes in the peripheral blood smear and a complex and life-threatening coagulation disorder causing recurrent venous thromboembolic events, severe thrombocytopenia, and subdural hematomas. Whole-exome sequencing revealed a frameshift mutation (C3AR1 c.355-356dup, p.Asp119Alafs*19) resulting in a premature stop in C3AR1 (Complement Component 3a Receptor 1). Based on this finding, atypical hemolytic uremic syndrome was suspected due to a genetic predisposition, and a targeted treatment regime with Eculizumab was initiated. Life-threatening hemostatic abnormalities would most likely have persisted had it not been for the implementation of whole-exome sequencing in this particular clinical setting. -

Diagnosis of Hemophilia and Other Bleeding Disorders
Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition Steve Kitchen Angus McCraw Marión Echenagucia Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2010 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. For permission to reproduce or translate this document, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel.: (514) 875-7944 Fax: (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition (2010) Steve Kitchen Angus McCraw Marión Echenagucia WFH Laboratory WFH Laboratory (co-author, Automation) Training Specialist Training Specialist Banco Municipal Sheffield Haemophilia Katharine Dormandy de Sangre del D.C. and Thrombosis Centre Haemophilia Centre Universidad Central Royal Hallamshire and Thrombosis Unit de Venezuela Hospital The Royal Free Hospital Caracas, Venezuela Sheffield, U.K. London, U.K. on behalf of The WFH Laboratory Sciences Committee Chair (2010): Steve Kitchen, Sheffield, U.K. Deputy Chair: Sukesh Nair, Vellore, India This edition was reviewed by the following, who at the time of writing were members of the World Federation of Hemophilia Laboratory Sciences Committee: Mansoor Ahmed Clarence Lam Norma de Bosch Sukesh Nair Ampaiwan Chuansumrit Alison Street Marión Echenagucia Alok Srivastava Andreas Hillarp Some sections were also reviewed by members of the World Federation of Hemophilia von Willebrand Disease and Rare Bleeding Disorders Committee. -

Approach to Bleeding Diathesi
Approach to Bleeding Diathesis Dr.Nalini K Pati MD, DNB, DCH (Syd), FRCPA Paediatric Haematologist Royal Children’s Hospital Melbourne Australia Objectives Objectives - I I. Clinical aspects of bleeding Clinical aspects of bleeding II. Hematologic disorders causing bleeding • Coagulation factor disorders • Platelet disorders III. Approach to acquired bleeding disorders • Hemostasis in liver disease • Surgical patients • Warfarin toxicity IV. Approach to laboratory abnormalities • Diagnosis and management of thrombocytopenia V. Drugs and blood products used for bleeding Clinical Features of Bleeding Disorders Petechiae Platelet Coagulation (typical of platelet disorders) disorders factor disorders Site of bleeding Skin Deep in soft tissues Mucous membranes (joints, muscles) (epistaxis, gum, vaginal, GI tract) Petechiae Yes No Ecchymoses (“bruises”) Small, superficial Large, deep Hemarthrosis / muscle bleeding Extremely rare Common Do not blanch with pressure Bleeding after cuts & scratches Yes No (cf. angiomas) Bleeding after surgery or trauma Immediate, Delayed (1-2 days), usually mild often severe Not palpable (cf. vasculitis) Ecchymoses (typical of coagulation factor disorders) Objectives - II Hematologic disorders causing bleeding – Coagulation factor disorders – Platelet disorders Coagulation factor disorders Hemophilia A and B Inherited bleeding Acquired bleeding Hemophilia A Hemophilia B disorders disorders Coagulation factor deficiency Factor VIII Factor IX – Hemophilia A and B – Liver disease – vonWillebrands disease – Vitamin K Inheritance X-linked X-linked recessive recessive – Other factor deficiencies deficiency/warfarin overdose Incidence 1/10,000 males 1/50,000 males –DIC Severity Related to factor level <1% - Severe - spontaneous bleeding 1-5% - Moderate - bleeding with mild injury 5-25% - Mild - bleeding with surgery or trauma Complications Soft tissue bleeding Hemarthrosis (acute) Hemophilia Clinical manifestations (hemophilia A & B are indistinguishable) Hemarthrosis (most common) Fixed joints Soft tissue hematomas (e. -

TPA-2012-2ING:Layout 1
106 Original Article DOI: 10.4274/tpa.823 Glanzmann thrombasthenia: Cerrahpaşa Medical Faculty experience Büşra Kutlubay, Gül Nihal Özdemir, Gülen Tüysüz, Hilmi Apak, Tiraje Celkan İstanbul University Cerrahpaşa Medical Faculty, Department of Pediatrics, Division of Pediatric Hematology-Oncology, İstanbul, Turkey Sum mary Aim: Glazmann thrombasthenia is a rare autosomal recessive disease characterized by a defect in platelet aggregation. Herein, we report the management of children with Glazmann thrombasthenia followed up at Cerrahpasa Medical Faculty Pediatric Hematology Department. Material and Method: The files of nineteen patients (42% girls, 58% boys; median age: 10 months) were retrospectively reviewed. Results: The median age of onset of bleeding symptoms was 9 months (2 weeks-24 months). All patients presented with easy bruising and mucosal bleeding. Fourteen patients’ parents were consanguineous. In 15 patients, flow cytometry was performed. According to this, 7 patients had type I, 6 patients had type II and 2 patients had type III disease. Nine patients were treated with thrombocyte transfusion, tranexamic acid, recombinant active factor VII and fibrin glue as a single or combined therapy; none of them had a major bleeding complication. Conclusions: Bleeding control during invasive procedures may be challenging in children with Glazmann thrombasthenia; local treatments, DDAVP, steroid and antifibrinolytics may be used with success. (Turk Arch Ped 2012; 47: 106-8) Key words: Glanzmann thrombasthenia, thrombocyte function disorder Introduction test, GT platelets do not aggregate following interaction with any substance except for ristocetin. In addition to Glanzmann thrombasthenia (GT) was described by Dr. aggregometer, low than normal levels of glycoprotein (GP) IIb- Eduard Glanzmann in 1918 for the first time as abnormal clot IIIa can be determined with flow cytometer using CD41 and retraction and defects in platelets (1). -

Dysfibrinogenemia and Thrombosis
Dys®brinogenemia and Thrombosis Timothy Hayes, DVM, MD c Objectives.ÐTo review the state of the art relating to sus of experts attending the conference was reached. The congenital dys®brinogenemia as a potential risk factor for results of the discussion were used to revise the manuscript thrombosis, as re¯ected by the medical literature and the into its ®nal form. consensus opinion of recognized experts in the ®eld, and Conclusions.ÐConsensus was reached on 5 conclusions to make recommendations for the use of laboratory assays and 2 recommendations concerning the use of testing for for assessing this thrombotic risk in individual patients. dys®brinogens in the assessment of thrombotic risk in in- Data Sources.ÐReview of the medical literature, pri- dividual patients. Detailed discussion of the rationale for marily from the last 10 years. each of these recommendations is found in the text of this Data Extraction and Synthesis.ÐAfter an initial assess- article. Compared with the other, more common heredi- ment of the literature, key points were identi®ed. Experts tary thrombophilias, dys®brinogenemia encompasses a di- were assigned to do an in-depth review of the literature verse group of defects with varied clinical expressions. and to prepare a summary of their ®ndings and recom- Congenital dys®brinogenemia is a relatively rare cause of mendations. A draft manuscript was prepared and circu- thrombophilia. Therefore, routine testing for this disorder lated to every participant in the College of American Pa- is not recommended as part of the laboratory evaluation thologists Conference on Diagnostic Issues in Thrombo- of a thrombophilic patient. -

Platelet Function Disorders
TREATMENT OF HEMOPHILIA APRIL 2008 • NO 19 PLATELET FUNCTION DISORDERS Second Edition Anjali A. Sharathkumar Amy Shapiro Indiana Hemophilia and Thrombosis Center Indianapolis, U.S.A. Published by the World Federation of Hemophilia (WFH), 1999; revised 2008. © World Federation of Hemophilia, 2008 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff.