Inheritance of Factor VII and Protein S Deficiency Together with Factor V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Protein C and S Deficiency in Deep Vein Thrombosis Patients Referred to Iranian Blood Transfusion Organization, Kermanshah
Protein C and S Deficiency in Deep Vein Thrombosis Patients Referred to Iranian Blood Transfusion Organization, Kermanshah Mehrdad Payandeh, 1 Mohammad Erfan Zare, 1, 2 Atefeh Nasir Kansestani, 1, 2 Kamran Ma nsouri, 1, 3 Zohreh Rahimi, 1, 4 Amir Hossein Hashemian, 5 Ebrahim Soltanian, 6 Hoshang Yousefi, 6 1Medical Biology Research Center, Kermanshah University of Medical Sciences, Kermanshah, Iran 2Student Research Committee, Kermanshah University of Medical Scien ces, Kermanshah, Iran 3Department of Molecular Medicine, School of advanced Medical Technologies, Tehran University of Medical Sciences, Tehran, Iran 4Department of Biochemistry, School of Medicine, Kermanshah University of Medical Sciences, Kermanshah, Ir an 5Department of Biostatistics, Faculty of Public Health, Kermanshah University of Medical Sciences, Kermanshah, Iran 6Research Center of Iranian Blood Transfusion Organization, Kermanshah, Iran Corresponding Author : Mohammad Erfan Zare, BSC student of M edical Lab Sciences. Medical Biology Research Center, P.O.Box: 1568, Sorkheh Lizheh, Kermanshah University of Medical Sciences, Kermanshah, Iran. E-mail : [email protected] Tel: +98 831 4276473 Fax: +98 831 4276471 Abstract Introduction: Normal homeostas is system has several inhibitor mechanisms in front of the amplifier’s natural clotting enzyme to prevent fibrin clots in the vessels. The main inhibitors of coagulation pathway are antithrombin (AT), protein C and protein S. Patients with hereditary defic iency of coagulation inhibitors are susceptible to venous thromboembolism (VTE). One of the major clinical manifestations of VTE is deep vein thrombosis (DVT). The present study has investigated the frequency of protein C and S deficiency among DVT patients that by using of these results and results from our previous study; we determined the most important hereditary risk factors for DVT in the Kermanshah Province of Iran with the Kurdish ethnic background. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -

Familial Haemophilia and Factor Vii Deficiency by M
J Clin Pathol: first published as 10.1136/jcp.11.5.412 on 1 September 1958. Downloaded from J. clin. Path. (1958), 11, 412. FAMILIAL HAEMOPHILIA AND FACTOR VII DEFICIENCY BY M. CONSTANDOULAKIS Front the Group Laboratory, St. Mary Abbots Hospital, London* (RECEIVED FOR PUBLICATION MAY 12, 1958) This investigation is reported because of the months later he was readmitted with a few bruises combination of familial haemophilia and factor on the legs and bleeding gums. Bleeding subsided with VII deficiency and the unusual occurrence of a local treatment and vitamin K1. female haemophiliac. Brenda, his sister (aged 10), appears normal with- Cases of combined deficiencies of different out any haemorrhagic manifestations up till now. factors are extremely rare and up till now Eileen Br., his mother (aged 46), revealed that she clotting had had excessive bleeding during both her deliveries, there have been reported combinations of haemo- during and after a hysterectomy, being transfused in philia and factor V deficiency (Koller, 1954), order to control haemorrhage, and after tooth extrac- haemophilia and Christmas disease (Soulier and tions at different times. Larrieu, 1953), Christmas disease and factor VII Frank G. (aged 43), his maternal uncle, has bled deficiency (Bell and Alton, 1955; Stein and since a small child after trauma and tooth extrac- Abrahams, 1956; de Vries, Kettenborg, and van tions, oozing from sockets usually persisting for about der Pol, 1955), but not of haemophilia and factor three days. In 1948 he had a blow in the abdomencopyright. VII deficiency. which was followed by severe haematemesis and Female haemophiliacs in the homozygous state melaena and he was transfused in order to control have been reported by Merskey (1951), Israels, the haemorrhage. -

Understanding Haemophilia
Understanding haemophilia Understanding haemophilia Contents Introduction 3 Haemophilia and your child 4 What is haemophilia? 5 What causes haemophilia? 5 Can females have haemophilia? 6 Carriers 8 Who is affected by haemophilia? 9 How severe is haemophilia? 9 Signs and symptoms of haemophilia 11 How is haemophilia diagnosed? 14 Diagnosis 14 Treatment 16 Port-a-cath 19 Managing joint bleeds with PRICE 19 Gene therapy 21 Possible complications of haemophilia 22 Inhibitors 22 Joint damage 22 Medical and dental treatment 23 Surgery Circumcision Dental care Medicines Vaccinations Bleeding disorder card Living with haemophilia 26 Sport and exercise 27 School, college and work 28 Travel 29 Pregnancy and haemophilia 30 Glossary of terms 32 About The Haemophilia Society 33 2 Understanding haemophilia Introduction This booklet is about haemophilia A and B. It gives a general overview of haemophilia and information on diagnosing, treating and living with the condition that we hope will answer your main questions. It has been written for people directly affected by haemophilia and for anyone interested in learning about haemophilia. If you are a parent and your child has recently been diagnosed with haemophilia you may be feeling quite overwhelmed. Remember, you’re not alone and many families are facing the same concerns and issues. Please do get in touch – we have lots of support and information available as well as services for parents and children. You can find out more via our website or Facebook pages, by emailing [email protected] or calling us on 020 7939 0780. The outlook is now the best it has ever been for people with haemophilia in the UK. -
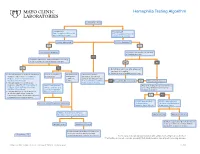
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further. -

Understanding Haemophilia CHAPTER 2
Understanding haemophilia CHAPTER 2 KEY POINTS • Haemophilia is an inherited condition caused by a gene alteration. • There are two types of haemophilia – A and B. • Haemophilia can be mild, moderate or severe. • Haemophilia is most commonly diagnosed in boys. • If you are considering having more children, there is support available to help with your decision. Haemophilia is an inherited bleeding disorder where blood doesn’t clot properly. It is caused when blood does not produce enough of one of the essential clotting ingredients. These ‘ingredients’ are clotting factors — proteins in the blood that control bleeding. The missing ingredient that causes haemophilia is usually either factor VIII (8) or IX (9). Roman numerals are used when referring to clotting factors. CHAPTER 2 2.1 UNDERSTANDING HAEMOPHILIA Blood clotting and bleeding Understanding how bleeding starts and stops NormalNormal clotting clotting process process Clotting factor activity Source: Hemophilia in Pictures. © WFH 2005. http://www1.wfh.org/publications/files/pdf-1311.pdf Bleeding starts when a capillary (small blood vessel) is injured and blood leaks out. When this happens, the capillary tightens up to slow the bleeding and blood cells called platelets make a plug to patch the hole. For people without haemophilia, the many clotting factors in plasma (part of the blood) knit together to make a clot over the plug. This makes the plug stronger and stops the bleeding. Clotting factor VIII and factor IX are essential to making the blood clot. 2.2 CHAPTER 2 UNDERSTANDING HAEMOPHILIA ClottingClotting in in haemophilia haemophilia Clotting factor activity Source: Hemophilia in Pictures. © WFH 2005. -

Factor Vii Deficiency/Mariani, Bernardi 401
Factor VII Deficiency Guglielmo Mariani, M.D.,1 and Francesco Bernardi, Ph.D.2 ABSTRACT The complex formed between the procoagulant serine protease activated factor VII (FVII) and the membrane protein tissue factor, exposed on the vascular lumen upon injury, triggers the initiation of blood clotting. This review describes the clinical picture of FVII deficiency and provides information on diagnosis and management of the disease. FVII deficiency, the most common among the rare congenital coagulation disorders, is transmitted with autosomal recessive inheritance. Clinical phenotypes range from asymp- tomatic condition, even in homozygotes, to severe disease characterized by life-threatening and disabling symptoms (central nervous system and gastrointestinal bleeding and hemarthrosis), with early age of presentation and the need for prophylaxis. In females, menorrhagia is prevalent and affects two thirds of the patients of fertile age. Although FVII gene mutations are extremely heterogeneous, several recurrent mutations have been reported, a few of them relatively frequent. The study of genotype-phenotype relationships indicates that modifier (environmental and/or inherited) components modulate expressiv- ity of FVII deficiency, as reflected by patients with identical FVII mutations and discordant clinical phenotypes. Several treatment options are available for FVII deficiency: the most effective are plasma-derived FVII concentrates and recombinant activated FVII (rFVIIa). Treatment-related side effects are rare. KEYWORDS: FVII deficiency, F7 database, -

Terminology Resource File
Terminology Resource File Version 2 July 2012 1 Terminology Resource File This resource file has been compiled and designed by the Northern Assistant Transfusion Practitioner group which was formed in 2008 and who later identified the need for such a file. This resource file is aimed at Assistant Transfusion Practitioners to help them understand the medical terminology and its relevance which they may encounter in the patient’s medical and nursing notes. The resource file will not include all medical complaints or illnesses but will incorporate those which will need to be considered and appreciated if a blood component was to be administered. The authors have taken great care to ensure that the information contained in this document is accurate and up to date. Authors: Jackie Cawthray Carron Fogg Julia Llewellyn Gillian McAnaney Lorna Panter Marsha Whittam Edited by: Denise Watson Document administrator: Janice Robertson ACKNOWLEDGMENTS We would like to acknowledge the following people for providing their valuable feedback on this first edition: Tony Davies Transfusion Liaison Practitioner Rose Gill Transfusion Practitioner Marie Green Transfusion Practitioner Tina Ivel Transfusion Practitioner Terry Perry Transfusion Specialist Janet Ryan Transfusion Practitioner Dr. Hazel Tinegate Consultant Haematologist Reviewed July 2012 Next review due July 2013 Version 2 July 2012 2 Contents Page no. Abbreviation list 6 Abdominal Aortic Aneurysm (AAA) 7 Acidosis 7 Activated Partial Thromboplastin Time (APTT) 7 Acquired Immune Deficiency Syndrome -

Delivery of Treatment for Haemophilia
WHO/HGN/WFH/ISTH/WG/02.6 ENGLISH ONLY Delivery of Treatment for Haemophilia Report of a Joint WHO/WFH/ISTH Meeting London, United Kingdom, 11 - 13 February 2002 Human Genetics Programme, 2002 Management of Noncommunicable Diseases World Health Organization Human Genetics Programme WHO/HGN/WFH/ISTH/WG/02.6 Management of Noncommunicable Diseases ENGLISH ONLY World Health Organization Delivery of Treatment for Haemophilia Report of a Joint WHO/WFH/ISTH Meeting London, United Kingdom, 11- 13 February 2002 Copyright ã WORLD HEALTH ORGANIZATION, 2002 All rights reserved. Publications of the World Health Organization can be obtained from Marketing and Dissemination, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 2476; fax: +41 22 791 4857; email: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to Publications, at the above address (fax: +41 22 791 4806; email: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned.