Approach to Bleeding Diathesi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Skin Injuries – Can We Determine Timing and Mechanism?
Skin injuries – can we determine timing and mechanism? Jo Tully VFPMS Seminar 2016 What skin injuries do we need to consider? • Bruising • Commonest accidental and inflicted skin injury • Basic principles that can be applied when formulating opinion • Abrasions • Lacerations }we need to be able to tell the difference • Incisions • Stabs/chops • Bite marks – animal v human / inflicted v ‘accidental’ v self-inflicted Our role…. We are often/usually/always asked…………….. • “What type of injury is it?” • “When did this injury occur?” • “How did this injury occur?” • “Was this injury inflicted or accidental?” • IS THIS CHILD ABUSE? • To be able to answer these questions (if we can) we need knowledge of • Anatomy/physiology/healing - injury interpretation • Forces • Mechanisms in relation to development, plausibility • Current evidence Bruising – can we really tell which bruises are caused by abuse? Definitions – bruising • BLUNT FORCE TRAUMA • Bruise =bleeding beneath intact skin due to BFT • Contusion = bruise in deeper tissues • Haematoma - extravasated blood filling a cavity (or potential space). Usually associated with swelling • Petechiae =Pinpoint sized (0.1-2mm) hemorrhages into the skin due to acute rise in venous pressure • medical causes • direct forces • indirect forces Medical Direct Indirect causes mechanical mechanical forces forces Factors affecting development and appearance of a bruise • Properties of impacting object or surface • Force of impact • Duration of impact • Site - properties of body region impacted (blood supply, -

Immune Thrombocytopenia Purpura (ITP)
Immune Thrombocytopenia Purpura (ITP) Information for patients and carers from the Haematology Department What is ITP? Immune thrombocytopenic purpura (ITP) is a condition which causes the number of platelets in Spleen your blood to be reduced. Platelets are cells that help blood to clot and they help to prevent bleeding and bruising after an injury. If you do not have enough platelets in your blood, you are likely to bruise easily or may be unable to stop bleeding if you cut yourself. In ITP, your body’s immune system destroys your own platelets. White blood cells in your blood and your spleen (an organ in your abdomen) are part of your immune system. One of their actions is to produce antibodies which help your body to fight Diagram showing the position of infections. If you develop ITP, your immune system the spleen becomes overactive and produces antibodies that cause your platelets to be destroyed in the spleen; this results in a low platelet count. ITP is a type of autoimmune condition (which means your immune system is acting against your body rather than for it). ITP in adults is more common in women than men. It is very different from ITP in children, who usually get ITP after a viral infection but who recover without any treatment. ITP in adults normally needs treatment. Some people with ITP have other autoimmune conditions, such as rheumatoid arthritis, or infections such as hepatitis or HIV. If you have any of these medical issues, your ITP may be treated slightly differently. 1 of 8 ITP (August 2021) A normal platelet count is between 150 and 400 thousand million platelets per litre of blood. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Bleeds and Bruises in Children with Haemophilia
Bleeds and Bruises in CHildren WiTH HaeMOPHilia MusCle ANd/or JoiNt Bleeds Call the parent/guardian P.r.i.C.e. siGNs oF A serious HeAd Bleed P : Protection * Headache. Lower Limb: Take weight off the joint or muscle * drowsiness. Upper Limb: No carrying using affected arm * Nausea. r : rest * Vomiting. • Rest means rest! * unsteady Balance. • Try not to allow use of the joint or muscle where * irritability. possible. * Confusion. * seizures. i : ice * loss of consciousness. • Regular ice packs can help with pain & reduce swelling. • Put an ice pack over the affected area for 20 minutes. Repeat every two hours. DO NOT leave the ice pack on for more than 20 minutes siGNs oF A soFt tissue DO NOT place ice pack directly on skin (Use a tea Bleed towel/cold pack cover) * Bruising, discolouring of skin. C : Compression * Mild swelling. • Use an elasticated bandage to compress the affected area to reduce swelling. e : elevation • Elevate the affected limb to help reduce swelling. siGNs oF AN ABdoMiNAl • Keep the affected joint or muscle above the level of the Bleed heart. * Bloody, black or tar-like First Aid bowel motions. * red or brown urine. Mouth & Gum Bleeds * Pain. These can be hard to control because clots that form are * Vomiting of blood (blood washed away by saliva or knocked off by the tongue or food. Try giving the child an ice cube or ice pop to suck. may be red or black). These bleeds may need treatment by parents or the treatment centre. Nosebleeds siGNs oF BleediNG iNto tHe Tilt head forward and pinch the bridge of the nose below the bone for 10 - 20 minutes and / or put an ice-pack on JoiNts or MusCles the bridge of the nose for not more than 5 minutes. -

Isolated Plantar Vein Thrombosis Resembling a Corn with a Bruise
JE Hahm, et al pISSN 1013-9087ㆍeISSN 2005-3894 Ann Dermatol Vol. 31, No. 1, 2019 https://doi.org/10.5021/ad.2019.31.1.66 CASE REPORT Isolated Plantar Vein Thrombosis Resembling a Corn with a Bruise Ji Eun Hahm, Kang Su Kim, Jae Won Ha, Chul Woo Kim, Sang Seok Kim Department of Dermatology, Kangdong Sacred Heart Hospital, College of Medicine, Hallym University, Seoul, Korea Plantar vein thrombosis, rarely-reported disease, is usually or callus, plantar fibromatosis, or plantar verruca1. Among accompanied by pain and tenderness in the plantar region laborers, they may develop from excess pressure on the and should be differentiated from other dermatological con- bony prominences of the feet, repetitive uneven friction ditions causing plantar pain, such as hemorrhagic corn/cal- from footwear, or gait abnormalities. Plantar vein throm- lus, plantar epidermal cyst, verruca, or plantar fibromatosis. bosis is a rare condition causing plantar pain. The exact A 52-year-old man presented with a violaceous tender sub- cause of plantar vein thrombosis is yet unclear, but predis- cutaneous nodule overlying a hyperkeratotic plaque on his posing conditions, such as prior trauma, surgery, paraneo- sole. Initially, he thought it was a corn and applied keratolytic plastic syndromes, or coagulation disorders have been agents, which failed to work. Sonography revealed a well-de- described. To date, there is no established treatment ex- marcated mass with increased peripheral vascularity. His cept surgical excision, but reportedly, nonsteroidal anti-in- pain was relieved after a complete wide excision, which con- flammatory drug or heparin with elastic bandage is known firmed the mass to be plantar vein thrombosis after histo- to be effective for symptomatic control2-5. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -

Understanding Haemophilia
Understanding haemophilia Understanding haemophilia Contents Introduction 3 Haemophilia and your child 4 What is haemophilia? 5 What causes haemophilia? 5 Can females have haemophilia? 6 Carriers 8 Who is affected by haemophilia? 9 How severe is haemophilia? 9 Signs and symptoms of haemophilia 11 How is haemophilia diagnosed? 14 Diagnosis 14 Treatment 16 Port-a-cath 19 Managing joint bleeds with PRICE 19 Gene therapy 21 Possible complications of haemophilia 22 Inhibitors 22 Joint damage 22 Medical and dental treatment 23 Surgery Circumcision Dental care Medicines Vaccinations Bleeding disorder card Living with haemophilia 26 Sport and exercise 27 School, college and work 28 Travel 29 Pregnancy and haemophilia 30 Glossary of terms 32 About The Haemophilia Society 33 2 Understanding haemophilia Introduction This booklet is about haemophilia A and B. It gives a general overview of haemophilia and information on diagnosing, treating and living with the condition that we hope will answer your main questions. It has been written for people directly affected by haemophilia and for anyone interested in learning about haemophilia. If you are a parent and your child has recently been diagnosed with haemophilia you may be feeling quite overwhelmed. Remember, you’re not alone and many families are facing the same concerns and issues. Please do get in touch – we have lots of support and information available as well as services for parents and children. You can find out more via our website or Facebook pages, by emailing [email protected] or calling us on 020 7939 0780. The outlook is now the best it has ever been for people with haemophilia in the UK. -
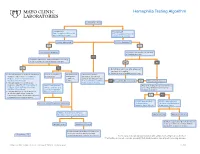
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further. -

ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4
ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4 ISTH 2012 11.6.2012 14:46 Page 1 Table of Contents 3 Welcome Message from the Meeting President 3 Welcome Message from ISTH Council Chairman 4 Welcome Message from SSC Chairman 5 Committees 7 ISTH Future Meetings Calendar 8 Meeting Sponsors 9 Awards and Grants 2012 12 General Information 20 Programme at a Glance 21 Day by Day Scientific Schedule & Programme 22 Detailed Programme Tuesday, 26 June 2012 25 Detailed Programme Wednesday, 27 June 2012 33 Detailed Programme Thursday, 28 June 2012 44 Detailed Programme Friday, 29 June 2012 56 Detailed Programme Saturday, 30 June 2012 68 Hot Topics Schedule 71 ePoster Sessions 97 Sponsor & Exhibitor Profiles 110 Exhibition Floor Plan 111 Congress Centre Floor Plan www.isth.org ISTH 2012 11.6.2012 14:46 Page 2 ISTH 2012 11.6.2012 14:46 Page 3 WelcomeCommittees Messages Message from the ISTH SSC 2012 Message from the ISTH Meeting President Chairman of Council Messages Dear Colleagues and Friends, Dear Colleagues and Friends, We warmly welcome you to the elcome It is my distinct privilege to welcome W Scientific and Standardization Com- you to Liverpool for our 2012 SSC mittee (SSC) meeting of the Inter- meeting. national Society on Thrombosis and Dr. Cheng-Hock Toh and his col- Haemostasis (ISTH) at Liverpool’s leagues have set up a great Pro- UNESCO World Heritage Centre waterfront! gramme aiming at making our off-congress year As setting standards is fundamental to all quality meeting especially attractive for our participants. -

Whole-Exome Sequencing of a Patient with Severe and Complex Hemostatic Abnormalities Reveals a Possible Contributing Frameshift Mutation in C3AR1
Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1 Eva Leinøe1, Ove Juul Nielsen1, Lars Jønson2 and Maria Rossing2∗ Department of Hematology1 and Center for Genomic Medicine2, Rigshospitalet, University of Copenhagen, Blegdamsvej 9, DK-2100 Copenhagen, Denmark Running head: WES reveals a C3AR1 mutation in a complex hemostatic patient ∗Corresponding author: Maria Rossing Center for Genomic Medicine Rigshospitalet University of Copenhagen Blegdamsvej 9 DK-2100 Copenhagen Denmark E-mail: [email protected] Phone: +45 3545 3016 Fax: +45 3545 4435 1 Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The increasing availability of genome-wide analysis has made it possible to rapidly sequence the exome of patients with undiagnosed or unresolved medical conditions. Here, we present the case of a 64-year-old male patient with schistocytes in the peripheral blood smear and a complex and life-threatening coagulation disorder causing recurrent venous thromboembolic events, severe thrombocytopenia, and subdural hematomas. Whole-exome sequencing revealed a frameshift mutation (C3AR1 c.355-356dup, p.Asp119Alafs*19) resulting in a premature stop in C3AR1 (Complement Component 3a Receptor 1). Based on this finding, atypical hemolytic uremic syndrome was suspected due to a genetic predisposition, and a targeted treatment regime with Eculizumab was initiated. Life-threatening hemostatic abnormalities would most likely have persisted had it not been for the implementation of whole-exome sequencing in this particular clinical setting.