In Ammatory Arthritis Complicating Galactosialidosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fast Urinary Screening of Oligosaccharidoses by MALDI-TOF/TOF Mass Spectrometry
Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry. Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, Charlotte Hinault To cite this version: Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, et al.. Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry.. Orphanet Journal of Rare Diseases, BioMed Central, 2014, 9 (1), pp.19. 10.1186/1750-1172-9-19. inserm- 00945684 HAL Id: inserm-00945684 https://www.hal.inserm.fr/inserm-00945684 Submitted on 12 Feb 2014 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Bonesso et al. Orphanet Journal of Rare Diseases 2014, 9:19 http://www.ojrd.com/content/9/1/19 RESEARCH Open Access Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry Laurent Bonesso1, Monique Piraud5, Céline Caruba1, Emmanuel Van Obberghen1,2,3,4, Raymond Mengual1† and Charlotte Hinault1,2,3,4*† Abstract Background: Oligosaccharidoses, which belong to the lysosomal storage diseases, are inherited metabolic disorders due to the absence or the loss of function of one of the enzymes involved in the catabolic pathway of glycoproteins and indirectly of glycosphingolipids. -

4Th Glycoproteinoses International Conference Advances in Pathogenesis and Therapy
Program & Abstracts 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ADVANCES IN PATHOGENESIS AND THERAPY ISMRD ST. LOUIS, MISSOURI, UNITED STATES Program & Abstracts I SM R D ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD would like to say A Very Special Thank You to the following organizations and companies who have very generously given donations and sponsorship to support the 4th International Conference on Glycoproteinoses THE PRENILLE EDWARD MALLINCKRODT FOUNDATION JR FOUNDATION MARK HASKINS I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD is very proud to display 10 featured Expression of Hope artworks to be Auctioned at the Gala Dinner. These beautiful prints are from Genzyme’s featured Artwork selection. Contents Welcome 1 SCIENTIFIC COMMITTEE: Stuart Kornfeld ISMRD Mission & Governance 3 (Chair, Scientifi c Planning Committee) Steve Walkley Sara Cathey ISMRD General Information 5 Richard Steet Sean Thomas Ackley, Philippines Miriam Storchli, Switzerland Alessandra d’Azzo ‘Hope’ by Sarah Noble, New Zealand Scientifi c Program 9 FAMILY CONFERENCE COMMITTEE: Family Program for Mucolipidosis 11 Jenny Noble (Conference Organiser) Jackie James (Conference Organiser Family Program For Alpha Mannosidosis /Sialidosis/ 13 - St. Louis) Fucosidosis/Aspartylglucosaminuria Mark Stark John Forman ‘All around the world’ by Zih Yun Li , Taiwan Childrens Program 16 Susan Kester Carolyn Paisley-Dew Tish Adkins Abstracts 17 Sara DeAngelis, Russia Gayle Rose, United States Speaker Profi les 60 Delegates 81 Helen Walker, Australia Nicklas Harkins, Canada Naomi Arai, Japan David Wentworth, Serbia I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts On behalf of the Scientifi c Planning Committee, I want to extend a warm welcome to all the investigators and Welcome! families who have traveled to St. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

The Lysosomal Sialic Acid Transporter Sialin Is Required for Normal CNS Myelination
The Journal of Neuroscience, December 9, 2009 • 29(49):15355–15365 • 15355 Neurobiology of Disease The Lysosomal Sialic Acid Transporter Sialin Is Required for Normal CNS Myelination Laura M. Prolo,1 Hannes Vogel,2 and Richard J. Reimer1 1Department of Neurology and Neurological Sciences and Graduate Program in Neuroscience and 2Departments of Pathology and Pediatrics, Stanford University School of Medicine, Stanford, California 94305 Salla disease and infantile sialic acid storage disease are autosomal recessive lysosomal storage disorders caused by mutations in the gene encoding sialin, a membrane protein that transports free sialic acid out of the lysosome after it is cleaved from sialoglycoconjugates undergoing degradation. Accumulation of sialic acid in lysosomes defines these disorders, and the clinical phenotype is characterized by neurodevelopmental defects, including severe CNS hypomyelination. In this study, we used a sialin-deficient mouse to address how loss of sialin leads to the defect in myelination. Behavioral analysis of the sialin ؊/؊ mouse demonstrates poor coordination, seizures, and premature death. Analysis by histology, electron microscopy, and Western blotting reveals a decrease in myelination of the CNS but normal neuronal cytoarchitecture and normal myelination of the PNS. To investigate potential mechanisms underlying CNS hypomyeli- nation, we studied myelination and oligodendrocyte development in optic nerves. We found reduced numbers of myelinated axons in optic nerves from sialin ؊/؊ mice, but the myelin that was present appeared grossly normal. Migration and density of oligodendrocyte precursorcellswerenormal;however,amarkeddecreaseinthenumberofpostmitoticoligodendrocytesandanassociatedincreaseinthe number of apoptotic cells during the later stages of myelinogenesis were observed. These findings suggest that a defect in maturation of cells in the oligodendrocyte lineage leads to increased apoptosis and underlies the myelination defect associated with sialin loss. -

Clinical, Biochemical, and Cytochemical Studies on a Japanese Salla Disease Case Associated with a Renal Disorder
J Hum Genet (2004) 49:656–663 DOI 10.1007/s10038-004-0203-y ORIGINAL ARTICLE Kouhei Ishiwari Æ Masaharu Kotani Æ Minoru Suzuki Elena Pumbo Æ Akemi Suzuki Æ Toshihide Kobayashi Tamaki Ueno Æ Tomoko Fukushige Æ Tamotsu Kanzaki Masato Imada Æ Kohji Itoh Æ Shinji Akioka Youichi Tajima Æ Hitoshi Sakuraba Clinical, biochemical, and cytochemical studies on a Japanese Salla disease case associated with a renal disorder Received: 27 July 2004 / Accepted: 6 September 2004 / Published online: 13 November 2004 Ó The Japan Society of Human Genetics and Springer-Verlag 2004 Abstract We report the first Japanese case of Salla dis- increased excretion of free sialic acid (N-acetylneurami- ease. A 5-year-old male patient developed unique pro- nic acid) into the patient’s urine. Immuno- and lectin teinuria with other clinical manifestations, including staining of the patient’s cells demonstrated the accu- coarse facies, dysostosis multiplex, mild mitral valve mulation of sialyl and asialyl glycoconjugates in lyso- regurgitation, umbilical and inguinal herniation, and somes and late endosomes. A defect in sialyl mild developmental delay. Pathological analysis of bi- glycoconjugate metabolism is thought to have occurred opsied kidney tissues showed marked vacuolation of in the patient’s cells, besides impairment of the lyso- podocytes, mesangial cells, capillary endothelial cells, somal transport of free sialic acid residues. A renal dis- and tubular cells. Biochemical studies involving thin- order should be considered as an important layer chromatography and mass spectrometry revealed manifestation, not only in infantile free sialic acid stor- age disease but also in Salla disease. K. Ishiwari Æ T. -

I-Cell Disease): a Rare Condition Resembling Hurler Syndrome: a Case Report S Yang, TW Yeung, HY Lau Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong
Hong Kong J Radiol. 2020;23:39-43 | https://doi.org/10.12809/hkjr2017142 CASE REPORT Mucolipidosis Type II (I-cell Disease): A Rare Condition Resembling Hurler Syndrome: A Case Report S Yang, TW Yeung, HY Lau Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong INTRODUCTION CASE REPORT Mucolipidosis type II (I-cell disease) is a rare autosomal A full term appropriate for gestational age recessive disorder of lysosomal metabolism with consanguineous Pakistani girl was born by emergency progressive multisystem deterioration that leads to Caesarean section for previous Caesarean section and death before or in early childhood. This disease was premature rupture of membrane. Antenatal history was first described in 1967 by Leroy and DeMars.1 Children unremarkable. There was a strong family history of with I-cell disease share many clinical and radiological I-cell disease. In the extended family, eight children had features with Hurler syndrome although there are distinct been diagnosed with the disease and an elder brother differences. Presentation of I-cell disease is earlier with a died from it at age 7 years. The patient presented with shorter clinical course, the radiological changes are more respiratory distress and was diagnosed to have transient profound, and the biochemical features are distinctive. tachypnoea of newborn. Initial chest radiograph The clinical course of I-cell disease is characterised revealed abnormal widened oar-shaped ribs (Figure 1). by progressive failure to thrive and developmental A skeletal survey -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Collectively Common, Individually Rare Many
OUTLOOK LYSOSOMAL STORAGE DISORDERS MYRIAD MALADIES u A tophag os om e Mutations affecting an es enzyme that labels other som do 3 enzymes for delivery to n the lysosome result in E 4 mucolipidosis-II Defects in a protein that helps endosomes and autophagosomes fuse with the lysosome cause Danon disease Defects in processing enzymes can cause Ly undigested material to Go soso lgi 2 me accumulate, such as in Gaucher’s disease and 5 Hunter syndrome En doplas mic retic ulum 1 Failure of processing in the endoplasmic reticulum can inac- 6 tivate a whole class Loss of transport proteins of enzymes, such as causes the lysosome to retain molecular building blocks in Nucleus in multiple sulfatase deficiency diseases such as cystinosis or sialic acid storage disease MANY MOVING PARTS The lysosome uses specialized enzymes to help cells to digest external biological materials and recycle defective proteins and damaged cellular machinery. Many LSDs arise from mutations in the genes that encode those enzymes, but there are numerous other ways in which this process can break down (red stars). 1 The endoplasmic The Golgi apparatus Endosomes transport The autophagosome Within the lysosome, Molecular building blocks reticulum performs the further processes and enzymes from the Golgi delivers damaged enzymes convert are released into cell for initial processing of labels specific enzymes and materials from organelles and misfolded molecules such as sugars, reuse newly synthesized for delivery to the outside the cell to the proteins to the lysosome proteins and lipids into lysosomal enzymes lysosome lysosome for recycling simpler building blocks COLLECTIVELY COMMON, INDIVIDUALLY RARE LSDs are not especially rare; estimates suggest that 1 in just over 5,000 newborns newborns worldwide, and the rarest have been described only a handful of times. -

Lysosomal Storage Disorders: Urine Screening
2460 Mountain Industrial Boulevard | Tucker, Georgia 30084 Phone: 470-378-2200 or 855-831-7447 | Fax: 470-378-2250 eglgenetics.com Lysosomal Storage Disorders: Urine Screening Test Code: BLSDS Turnaround time: 2 weeks (GAGs performed Fri 10 am / Oligos performed Fri 3 pm) CPT Codes: 82542 x1, 82570 x1, 83864 x1, 84275 x1, 84375 x1, 84377 x1 Condition Description Notice: This test has been discontinued EGL no longer accepts samples for this test. For questions, please call: 470-378-2200 In glycoprotein storage diseases (GSDs), certain subtypes of congenital disorders of glycosylation (CDGs), and in the mucolipidoses, there is an accumulation of oligosaccharides, free glycans, glycoamino acids, glycolipid and glycopeptide in the urine. Glycoprotein storage diseases are genetic conditions caused by the body's inability to produce specific enzymes. Normally, the body uses enzymes to process, break down and recycle materials in cells. In individuals with GSD and related diseases, the missing or insufficient enzyme prevents the proper processing and recycling process. This results in the storage of materials, called oligosaccharides or free glycans and glycoamino acids in virtually every cell of the body. As a result, cells do not perform properly and may cause progressive damage throughout the body, including the heart, bones, joints, respiratory system, immune system and central nervous system. While the disease may or may not be apparent at birth, signs and symptoms develop with age as more cells become damaged by the accumulation of materials. The symptoms of these diseases may vary based on syndrome type, and in some cases may resemble a mucopolysaccharidosis. This urinary oligosaccharide and glycan screening uses mass spectrometry (MS), which provides a better sensitivity and specificity than traditional TLC methods. -

Conventional and Unconventional Therapeutic Strategies for Sialidosis Type I
Journal of Clinical Medicine Article Conventional and Unconventional Therapeutic Strategies for Sialidosis Type I 1, 1, 1 1,2 Rosario Mosca y, Diantha van de Vlekkert y , Yvan Campos , Leigh E. Fremuth , Jaclyn Cadaoas 3 , Vish Koppaka 3, Emil Kakkis 3, Cynthia Tifft 4, Camilo Toro 5 , Simona Allievi 6,7, Cinzia Gellera 6,7, Laura Canafoglia 7 , Gepke Visser 8 , Ida Annunziata 1 and Alessandra d’Azzo 1,* 1 Department of Genetics, St. Jude Children’s Research Hospital, Memphis, TN 38105, USA; [email protected] (R.M.); [email protected] (D.v.d.V.); [email protected] (Y.C.); [email protected] (L.E.F.); [email protected] (I.A.) 2 Department of Anatomy and Neurobiology, College of Graduate Health Sciences, University of Tennessee Health Science Center, Memphis, TN 38163, USA 3 Ultragenyx Pharmaceutical, Novato, CA 94949, USA; [email protected] (J.C.); [email protected] (V.K.); [email protected] (E.K.) 4 Office of the Clinical Director & Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health (NHGRI), Bethesda, MD 20892, USA; [email protected] 5 Undiagnosed Disease Network, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] 6 Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy; [email protected] (S.A.); [email protected] (C.G.) 7 Neurophysiopathology, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy; [email protected] 8 Department of Metabolic Diseases, Wilhelmina Children’s Hospital, University Medical Center Utrecht, 3584 CX Utrecht, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +1-901-595-2698 These authors contributed equally to this work. -
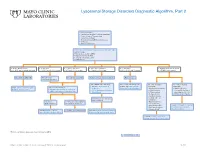
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2 Clinical information: ■ Developmental delay/Cognitive impairment ■ Coarse features/Organomegaly ■ Dysostosis multiplex ■ Neurodegeneration/Behavioral changes ■ Ichthyosis ■ Hearing defects/loss LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: ML II/III profile ■ S: abnormal ■ CT and S: abnormal ■ MPS and S: abnormal ■ CT: abnormal ■ OLIGO: characteristic profile ■ CT, MPS and S: normal/abnormal ■ CT, MPS and OLIGO: normal ■ MPS and OLIGO: normal ■ CT and OLIGO: normal ■ MPS, OLIGO and S: normal ■ CT, MPS and S; normal Mucolipidosis (ML) II/III Metachromatic Prosaposin/ SaposinB Multiple sulfatase deficiency (MSD) Fabry disease leukodystrophy (MLD) Order BOTH of the following: For recommended diagnostic One of the following One of the following ■ ARSAW / Arylsulfatase A, workup, see Fabry Disease suspected: suspected: Order: GNPTZ / GNPTAB Gene, Order 1 of the following: Leukocytes Diagnostic Testing Algorithm ■ Aspartylglucosaminuria ■ NGLY1 deficiency Full Gene Analysis, Varies ■ ARSAW / Arylsulfatase A, Leukocytes ■ I2SW / Iduronate-2-Sulfatase, ■ α-Mannosidosis (Congenital disorder of ■ ARSU / Arylsulfatase A, 24 Hour, Urine Whole Blood ■ β-Mannosidosis glycosylation: CDG-Iv) ■ Pompe disease ■ MOGS-CDG (CDG-IIb) ■ Sandhoff disease ■ Schindler disease If both results are deficient, ■ Sialidosis MSD confirmed If deficient, If normal, possible Saposin -

Novel Missense Mutations in the Human Lysosomal Sialidase Gene in Sialidosis Patients and Prediction of Structural Alterations of Mutant Enzymes
B.J Hum Jochimsen Genet et(2002) al.: Stetteria 47:29–37 hydrogenophila © Jpn Soc Hum Genet and Springer-Verlag4600/29 2002 ORIGINAL ARTICLE Kohji Itoh · Yasunori Naganawa · Fumiko Matsuzawa Seiichi Aikawa · Hirofumi Doi · Naokazu Sasagasako Takeshi Yamada · Jun-ichi Kira · Takuro Kobayashi Alexey V. Pshezhetsky · Hitoshi Sakuraba Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes Received: Stptember 21, 2001 / Accepted: November 2, 2001 Abstract Three novel missense mutations in the human changes including the active site residues responsible for lysosomal sialidase gene causing amino acid substitutions binding the sialic acid carboxylate group. The W240R sub- (P80L, W240R, and P316S) in the coding region were stitution was deduced to influence the molecular surface identified in two Japanese sialidosis patients. One patient structure of a limited region of the constructed models, with a severe, congenital form of type 2 sialidosis was a which was also influenced by previously identified V217M compound heterozygote for 239C-to-T (P80L) and 718T-to- and G243R transversions. C (W240R). The other patient with a mild juvenile-onset phenotype (type 1) was a homozygote for the base substitu- Key words Lysosomal sialidase · Sialidosis · Molecular tion of 946C-to-T (P316S). None of these mutant cDNA modeling · Protective protein/cathepsin A · Galacto- products showed enzymatic activity toward an artificial sialidosis substrate when coexpressed in galactosialidosis fibroblastic cells together with protective protein/cathepsin A (PPCA). All mutants showed a reticular immunofluorescence distri- bution when coexpressed with the PPCA gene in COS-1 Introduction cells, suggesting that the gene products were retained in the endoplasmic reticulum/Golgi area or rapidly degraded Lysosomal sialidase (neuraminidase, EC 3.2.1.18) catalyzes in the lysosomes.