Steps Toward the Synthesis of 1,1'-Dideaza- Quinine: A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies Toward Synthesis of Polycyclic Polyprenylated Acylphloroglucinols
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Roxana Ciochina University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ciochina, Roxana, "STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS" (2006). University of Kentucky Doctoral Dissertations. 291. https://uknowledge.uky.edu/gradschool_diss/291 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Roxana Ciochina The Graduate School University of Kentucky 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Sciences at the University of Kentucky By Roxana Ciochina Lexington, KY Director: Dr. R. B. Grossman, Professor of Chemistry Lexington, KY 2006 ABSTRACT OF DISSERTATION STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of compounds that reveal intriguing biological activities and interesting and challenging chemical structures. These products are claimed to possess antioxidant, antiviral, and antimitotic properties. Increasing interest is related to their function in the CNS as modulators of neurotransmitters associated to neuronal damaging and depression. All these features make PPAPs targets for synthesis. We decided to focus our own initial efforts in this area on the type A PPAP, nemorosone because we thought that its fairly simple structure relative to other PPAPs would present fewer hurdles as we developed our methodology. -

Synthesis of Alkynyl Ribofuranosides
City University of New York (CUNY) CUNY Academic Works Dissertations and Theses City College of New York 2011 Synthesis of Alkynyl Ribofuranosides Christian Rodriguez CUNY City College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/cc_etds_theses/25 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] SYNTHESIS OF ALKYNYL RIBOFURANOSIDES A Thesis Presented to The Faculty of the Chemistry Program The City College of New York In (Partial) Fulfillment of the Requirements for the Degree Master of Arts by Christian Rodriguez December, 2010 - 1 - Synthesis of Alkynyl Ribofuranosides By Christian Rodriguez Mentor: P. Meleties Table of Contents Chapter 1 1.1 Introduction 6 1.2 Preparation of ribonolactone template 8 1.3 Synthesis of protected ribonolactone 9 Chapter 2 2.1 Preparation of ethynyl ribofuranosides 10 2.2 Reaction with ethynylmagnesium bromide 12 2.3 Intramolecular cyclization of diyne diol 14 2.4 Reaction of ribonolactone with lithium acetylide 15 2.5 Boron trifluoride hemiacetal deoxygenation 15 2.6 Alternative deacetylation 16 Chapter 3 3.1 Selecting appropriate protecting group 19 3.2 Preparation of trimethylsilyl alkynyl 5-O-benzyl-2,3-O isopropylidene 19 ribofuranoside 3.3 Lewis acid promoted triethylsilane dehydroxylation mechanism 20 Chapter 4 4.1 Hemiacetal alkynylation 25 4.2 Hemiacetal nucleophilic addition mechanism 26 4.3 Intramolecular -
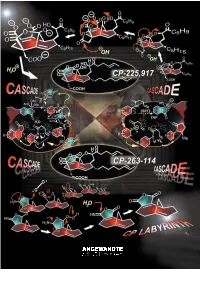
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

The Claisen Condensation
Lecture 19 The Claisen Condensation •• •• • • O • O • – • CH3COCH2CH3 • CH2 COCH2CH3 March 29, 2016 Chemistry 328N Exam Tomorrow Evening!! Review tonight 5PM -6PM Welch 1.316 Chemistry 328N Some “loose ends” before we go on Spectrosopy of acid derivatives A selective reduction for your tool box Chemistry 328N Reduction of Acid Derivatives Acids (page 679-681) Esters (page 738-739) Please work through the example on 738 Amides (page 739-742) Nitriles (page 742) Selective reductions with NaBH4 Chemistry 328N DIBAlH Diisobutylaluminum hydride (DIBAlH) at -78°C selectively reduces esters to aldehydes •at -78°C, the tetrahedral intermediate does not collapse and it is not until hydrolysis in aqueous acid that the carbonyl group of the aldehyde is liberated Stable at low temperature Chemistry 328N Infrared Spectroscopy C=O stretching frequency depends on whether the compound is an acyl chloride, anhydride, ester, or amide. C=O stretching frequency n O O O O O CH3CCl CH3COCCH3 CH3COCH3 CH3CNH2 1822 cm-1 1748 1736 cm-1 1694 cm-1 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Anhydrides have two peaks due to C=O stretching. One from symmetrical stretching of the C=O and the other from an antisymmetrical stretch. C=O stretching frequency n O O CH3COCCH3 1748 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Nitriles are readily identified by absorption due to carbon-nitrogen triple bond stretching that is “all alone” in the 2210-2260 cm-1 region. Chemistry 328N Hydrolysis and Decarboxylation Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl ester hydrolysis Note which bond is broken in this hydrolysis !! Chemistry 328N Recall our discussion of the acidity of protons a to carbonyls The anion is stabilized by resonance The better the stabilization, the more acidic the a proton Acidity of a protons on“normal” aldehydes and ketones is about that of alcohols and less than water…pKa ~ 18-20 Some are far more acidic, i.e. -

The Claisen Condensation Is a Carbon–Carbon Bond Forming Reaction That Occurs Between Two Esters Or One Ester and Another Carb
Claisen Ester Condensation The Claisen condensation is a carbon–carbon bond forming reaction that occurs between two esters or one ester and another carbonyl compound in the presence of a strong base, resulting in a β-keto ester or a β-diketone. Requirements ❖ At least one of the reagents must be enolizable (have an α-proton and be able to undergo deprotonation to form the enolate anion). ❖ There are a number of different combinations of enolizable and nonenolizable carbonyl compounds that form a few different types of Claisen. ❖ The base used must not interfere with the reaction by undergoing nucleophilic substitution or addition with a carbonyl carbon. ❖ For this reason, the conjugate sodium alkoxide base of the alcohol formed (e.g. sodium ethoxide if ethanol is formed) is often used, since the alkoxide is regenerated. ❖ In mixed Claisen condensations, a non-nucleophilic base such as lithium diisopropylamide, or LDA, may be used, since only one compound is enolizable. ❖ LDA is not commonly used in the classic Claisen or Dieckmann condensations due to enolization of the electrophilic ester. ❖ The alkoxy portion of the ester must be a relatively good leaving group. ❖ Methyl and ethyl esters, which yields methoxide and ethoxide, respectively, are commonly used. Types Mechanism ❖ In the first step of the mechanism, an α-proton is removed by a strong base, resulting in the formation of an enolate anion, which is made relatively stable by the delocalization of electrons. ❖ Next, the carbonyl carbon of the (other) ester is nucleophilically attacked by the enolate anion. ❖ The alkoxy group is then eliminated (resulting in (re)generation of the alkoxide), and the alkoxide removes the newly formed doubly α-proton to form a new, highly resonance-stabilized enolate anion. -

Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B
J. Am. Chem. Soc. 2000, 122, 10033-10046 10033 Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B David A. Evans,* Duke M. Fitch,1 Thomas E. Smith, and Victor J. Cee Contribution from the Department of Chemistry and Chemical Biology, HarVard UniVersity, Cambridge, Massachusetts 02138 ReceiVed June 29, 2000 Abstract: The synthesis of phorboxazole B has been accomplished in 27 linear steps and an overall yield of 12.6%. The absolute stereochemistry of the C4-C12,C33-C38, and C13-C19 fragments was established utilizing catalytic asymmetric aldol methodology, while the absolute stereochemistry of the C20-C32 fragment was derived from an auxiliary-based asymmetric aldol reaction. All remaining chirality was incorporated through internal asymmetric induction, with the exception of the C43 stereocenter which was derived from (R)-trityl glycidol. Key fragment couplings include a stereoselective double stereodifferentiating aldol reaction, a metalated oxazole alkylation, an oxazole-stabilized Wittig olefination, and a chelation-controlled addition of the fully elaborated alkenyl metal side chain. Introduction and HT29 (3.31 × 10-10 M), as well as leukemia CCRF-CBM (2.45 × 10-10 M), prostate cancer PC-3 (3.54 × 10-10 M), and Phorboxazoles A (1)andB(2) are marine natural products breast cancer MCF7 cell lines (5.62 × 10-10 M).2b In addition isolated from a recently discovered species of Indian Ocean to this impressive anticancer activity, phorboxazoles A and B sponge (genus Phorbas sp.) near Muiron Island, Western also exhibit potent in vitro antifungal activity against Candida Australia.2 These substances are representative of a new class albicans and Saccharomyces carlsbergensis at 0.1 µg/disk in of macrolides differing only in their C hydroxyl-bearing 13 the agar disk diffusion assay.2a stereocenters. -

Use of the Non-Aldol Aldol Process in the Synthesis of the C1–C11 Fragment of the Tedanolides: Use of Lactol Ethers in Place of Tetrahydrofurans
TETRAHEDRON LETTERS Pergamon Tetrahedron Letters 41 (2000) 9719–9723 Use of the non-aldol aldol process in the synthesis of the C1–C11 fragment of the tedanolides: use of lactol ethers in place of tetrahydrofurans Michael E. Jung* and Christopher P. Lee Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095-1569, USA Received 15 August 2000; accepted 18 September 2000 Abstract The use of a lactol methyl ether 23 in place of the simple tetrahydrofuran 11 allows for the high yielding non-aldol aldol process to occur without concomitant tetrahydropyran formation (cf. 13) to give the desired product 24 in good yield. © 2000 Elsevier Science Ltd. All rights reserved. Keywords: non-aldol aldol; polypropionate synthesis; lactol methyl ethers. Tedanolide (1,R=OH) was isolated by Schmitz and co-workers in 1984 from the Caribbean 1 sponge Tedania ignis. The macrolide demonstrates its high cytotoxicity by displaying ED50’s of 250 pg/mL against human nasopharynx carcinoma and 16 pg/mL against in vitro lymphocytic leukemia. Seven years after tedanolide’s discovery, Fusetani and co-workers isolated 13-deoxy- tedanolide (2,R=H) from the Japanese sponge Mycale adhaerens.2 This macrolide is also extremely cytotoxic, exhibiting an IC50 of 94 pg/mL against P388 murine leukemia. Due to its powerful antitumor activity and complex structure, tedanolide has garnered considerable synthetic interest,3 including that of our group which uses the non-aldol aldol process.4 Disconnecting the tedanolide backbone retrosynthetically is quite straightforward, beginning with cleavage at the lactone moiety and at the C12C13 bond, which could be formed in the forward sense by an aldol reaction for 1 or an alkylation for 2, either prior to a macrolactoniza- tion or after simple ester formation. -

II Reduction Reactions
II Reduction Reactions Objectives By the end of this section you will: 1) be able to exploit the differences in reactivity of various reducing agents (hydride vs neutral reductants) in chemoselective reductions and be able to provide a mechanistic rationale to account for their differing reactivities; 2) be able to use the inherent chirality in a substrate to control the outcome of a reduction of proximal ketones to generate selectively syn and anti 1,3- and 1,2-diols; 3) be able to rationalise the outcome of these diastereoselective reactions using T.S. diagrams; 4) have gained an appreciation of the versatility of transition metals in reduction reactions; 5) have gained an appreciation of the synthetic utility of dissolving metal reductions; 6) be able to use radical chemistry for deoxygenation and reduction of halides. II.A Reduction of Carboxylic Acid Derivatives and Related Functionality OR' H ROH RO RO carboxylic acid aldehyde primary alcohol derivatives R N RNH2 RNO2 Issues of Reactivity and Selectivity Similar issues of selectivity and reactivity to those we encountered in the case of oxidation reactions also arise in reduction reactions. 1. Chemoselectivity. Many different functional groups can be reduced in a variety of ways. We often need to selectively reduce one functional group whilst leaving others intact (remember year 1 practical!). NaBH4 Sn, HCl OH O O O2N O2N H2N Chemoselective reductions from a practical in CHM1C3 2. In the case of carboxylic acid derivatives there are two possible reduction products: an aldehyde and an alcohol. Ideally we need methods for selectively accessing either product. -

Studies on the Prebiotic Origin of 2-Deoxy-D-Ribose
Studies on the Prebiotic Origin of 2-Deoxy-D-ribose Andrew Mark Steer Doctor of Philosophy University of York Chemistry August 2017 Abstract DNA is an important biological structure necessary for cell proliferation. The origins of cell- like structures and the building blocks of DNA are therefore also of great concern. As of yet the prebiotic origin of 2-deoxy-D-ribose, the sugar of DNA, has no satisfactory explanation. This research attempts to provide a possible explanation to the chemical origin of 2-deoxy- D-ribose via an aldol reaction between acetaldehyde 1 and D-glyceraldehyde D-2 (Error! Reference source not found.). The sugar mixture is trapped with N,N-diphenylhydrazine 3 for ease of purification and characterisation. The reaction is promoted by amino acids, amino esters and amino nitriles consistently giving selectivities in favour of 2-deoxy-D- ribose. This is the first example of an amino nitrile promoted reaction. Potential prebiotic synthesis of 2-deoxy-D-ribose and subsequent trapping with N,N-diphenyl hydrazine 3. The research is developed further by exploring the formation of 2-deoxy-D-ribose in a “protocell” environment – a primitive cell. Here we suggest that primitive cells may have been simple hydrogel systems. A discussion of the characterisation and catalytic ability of small peptide-based supramolecular structures is included. ii Contents Abstract ............................................................................................................................ ii Contents ......................................................................................................................... -

Efforts Towards the Total Synthesis of Azaspiracid-3 DISSERTATION
Efforts towards the Total Synthesis of Azaspiracid-3 DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Zhigao Zhang Graduate Program in Chemistry The Ohio State University 2013 Dissertation Committee: Dr. Craig J. Forsyth, Advisor Dr. Jon R. Parquette Dr Anita E. Mattson Copyright by Zhigao Zhang 2013 Abstract Azaspiracid, a structurally complex marine toxin isolated from the mussel Mytilus edulis in Killary Harbor, Ireland, represents a new class of marine metabolites unrelated to any previous known agent of diarrhetic shellfish posioning. As such, the natural product has spurred considerable interest among organic community. The thesis details the study towards the total synthesis of azaspiracid-3. The C1-C21 domain and C22-C40 domain have been synthesized, which aims to assemble the molecule through Yamaguchi esterification. The ABCD domain involved an efficient NHK coupling reaction between C1-C12 segment and C13-C22 segment. The trioxadispiroketal skeleton was constructed under the influence of acid catalysis. The EFGHI domain highlighted a chelate controlled Mukaiyama aldol reaction to produce C22-C40 linear precursor, and a DIHMA process to assemble the bridged ketal. With the recognition of bridged ketal and spiroaminal functionality in the FGHI domain, the gold-catalysis to achieve these moieties has been proposed. A novel gold- catalyzed spiroaminal formation has been systemically developed. The gold-catalyzed ketallization was then successfully implemented to construct the bridged ketal, while an inevitable elimination could not deliver the desired spiroaminal under gold conditions. A NHK reaction to couple the C1-C21 and C22-C40 domains has also been proposed. -

CHEM 330 Topics Discussed on Sept. 18 Principle: a Claisen Condensation Promoted by Nah/Cat. Etoh Takes Place So That All Steps
CHEM 330 Topics Discussed on Sept. 18 Principle: a Claisen condensation promoted by NaH/cat. EtOH takes place so that all steps but the final deprotonation of EtOH by NaH are reversible. Consequently, the system will tend to attain a thermodynamic energy minimum, and the product that forms will be the thermodynamically most favorable one. A reaction occurring under such conditions is said to proceed under thermodynamic control Principle: mixing one equivalent of pure ethyl acetoacetate with excess pure ethanol in the presence of a catalytic amount of EtONa will induce a reverse Claisen condensation ("retro-Claisen"), resulting in conversion of the starting acetoacetate into two molecules of ethyl acetate (the thermodynamically more favorable state of the system): – CH3-CO-CH2-COOEt + EtOH —[ cat. EtO ]—> 2 CH3-COOEt (overall ΔG < 0) Principle of microscopic reversibility: in each step of any given transformation, the forward and the reverse reactions occur along the same mechanistic pathway; i.e., by identical mechanisms operating in reverse Example: a reverse Claisen condensation occurs as follows: O O O O Na + Na + H–OEt OEt OEt OEt pKa ≈ 12 H pKa ≈ 17 + H–OEt + H–OEt 5 Keq ≈ 10 although the above equilibrium is shifted to the right, there will always be some residual free EtO and ethyl acetoacetate in the medium. Under the usual approximations (CHEM 203): –5 –3 [CH3COCH2COOEt] = [EtO ] ≈ 10 ≈ 3•10 – These residual concentrations of EtO( ) and CH3COCH2COOEt are sufficient to induce a slow, but inexorable, reverse reaction. O O O O O O + Na OEt OEt OEt OEt OEt OEt + H–OEt + H–OEt Na + H–OEt O O OEt OEt OEt Na H notice that the reversible reaction depends on proton exchanges among the various species: NaH removes all such protons and suppresses the retro-Claisen process lecture of Sept 18 p. -

Nucleophilic Addition to the Carbonyl Group 6
Nucleophilic addition to the carbonyl group 6 Connections Building on Arriving at Looking forward to • Functional groups, especially the C=O • How and why the C=O group reacts • Additions of organometallic group ch2 with nucleophiles reagents ch9 • Identifying the functional groups in a • Explaining the reactivity of the C=O • Substitution reactions of the C=O molecule spectroscopically ch3 group using molecular orbitals and curly group’s oxygen atom ch11 • How molecular orbitals explain arrows • How the C=O group in derivatives of molecular shapes and functional • What sorts of molecules can be made by carboxylic acids promotes substitution groups ch4 reactions of C=O groups reactions ch10 • How, and why, molecules react together • How acid or base catalysts improve the • C=O groups with an adjacent double and using curly arrows to describe reactivity of the C=O group bond ch22 reactions ch5 Molecular orbitals explain the reactivity of the carbonyl group We are now going to leave to one side most of the reactions you met in the last chapter—we will come back to them all again later in the book. In this chapter we are going to concentrate on just one of them—probably the simplest of all organic reactions—the addition of a nucleo- phile to a carbonyl group. The carbonyl group, as found in aldehydes, ketones, and many other compounds, is without doubt the most important functional group in organic chemis- try, and that is another reason why we have chosen it as our fi rst topic for more detailed study. You met nucleophilic addition to a carbonyl group on pp.