Efforts Towards the Total Synthesis of Azaspiracid-3 DISSERTATION
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules
ACCOUNT 21 Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules Shunsuke Chiba* Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore Fax +6567911961; E-mail: [email protected] Received 31 May 2012 Organic azides possess diverse chemical reactivities.4 Abstract: In this account, recent advances made on the reactions of several types of organic azides, such as vinyl azides, cyclic 2-azido Owing to their 1,3-dipole character, they undergo [3+2] alcohols, a-azido carbonyl compounds, towards the synthesis of cycloaddition with unsaturated bonds, such as those in nitrogen-containing molecules are described. alkynes and alkenes as well as carbonitriles (Scheme 1, part a).5 Organic azides can also be regarded as nitrene 1 Introduction equivalents (Scheme 1, part b).6 Accordingly, their reac- 2 Chemistry of Vinyl Azides tions with nucleophilic anions, electrophilic cations, and 2.1 Thermal [3+2]-Annulation of Vinyl Azides with 1,3-Dicar- radicals can formally provide the corresponding nitrogen bonyl Compounds 2.2 Manganese(III)-Catalyzed Formal [3+2]-Annulation with anions, cations, and radicals, respectively, forming a new 1,3-Dicarbonyl Compounds bond with the internal azido nitrogen and releasing molec- 2.3 Manganese(III)-Mediated/Catalyzed Formal [3+3]-Annu- ular nitrogen. Moreover, the generation of anions, cations, lation with Cyclopropanols and radicals at the a-position to the azido moiety can re- 2.4 Synthesis of Isoquinolines from a-Aryl-Substituted Vinyl sult in rapid denitrogenation to deliver the corresponding Azides and Internal Alkynes by Rhodium–Copper Bimetal- iminyl species, which can be used in further synthetic lic Cooperation transformations (i.e., carbon–nitrogen bond formation). -

Studies Toward Synthesis of Polycyclic Polyprenylated Acylphloroglucinols
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Roxana Ciochina University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ciochina, Roxana, "STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS" (2006). University of Kentucky Doctoral Dissertations. 291. https://uknowledge.uky.edu/gradschool_diss/291 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Roxana Ciochina The Graduate School University of Kentucky 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Sciences at the University of Kentucky By Roxana Ciochina Lexington, KY Director: Dr. R. B. Grossman, Professor of Chemistry Lexington, KY 2006 ABSTRACT OF DISSERTATION STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of compounds that reveal intriguing biological activities and interesting and challenging chemical structures. These products are claimed to possess antioxidant, antiviral, and antimitotic properties. Increasing interest is related to their function in the CNS as modulators of neurotransmitters associated to neuronal damaging and depression. All these features make PPAPs targets for synthesis. We decided to focus our own initial efforts in this area on the type A PPAP, nemorosone because we thought that its fairly simple structure relative to other PPAPs would present fewer hurdles as we developed our methodology. -

Oxidation of Secondary Alcohols to Ketones
Oxidation of secondary alcohols to ketones The oxidation of secondary alcohols to ketones is an important oxidation reaction in organic chemistry. Where a secondary alcohol is oxidised, it is converted to a ketone. The hydrogen from the hydroxyl group is lost along with the hydrogen bonded to the second carbon. The remaining oxygen then forms double bonds with the carbon. This leaves a ketone, as R1–COR2. Ketones cannot normally be oxidised any further because this would involve breaking a C–C bond, which requires too much energy.[1] The reaction can occur using a variety of oxidants. Contents Potassium dichromate PCC (Pyridinium chlorochromate) Dess–Martin oxidation Swern oxidation Oppenauer oxidation Fétizon oxidation See also References Potassium dichromate A secondary alcohol can be oxidised into a ketone using acidified potassium dichromate and heating under 2− 3+ reflux. The orange-red dichromate ion, Cr2O7 , is reduced to the green Cr ion. This reaction was once used in an alcohol breath test. PCC (Pyridinium chlorochromate) PCC, when used in an organic solvent, can be used to oxidise a secondary alcohol into a ketone. It has the advantage of doing so selectively without the tendency to over-oxidise. Dess–Martin oxidation The Dess–Martin periodinane is a mild oxidant for the conversion of alcohols to aldehydes or ketones.[2] The reaction is performed under standard conditions, at room temperature, most often in dichloromethane. The reaction takes between half an hour and two hours to complete. The product is then separated from the spent periodinane.[3] Swern oxidation Swern oxidation oxidises secondary alcohols into ketones using oxalyl chloride and dimethylsulfoxide. -

Safe Handling of Sodium Azide (SAZ)
Safe Handling of Sodium Azide (SAZ) 1,2 Sodium azide (SAZ, CAS# 26628-22-8) is a white crystalline solid [molecular formula of (NaN3)] used in organic synthesis and also as a well-known preservative at low concentrations in molecular biology reagents. Azide chemistry3,4 offers an effective means to synthesize a range of nitrogen-containing compounds with a wide variety of functional groups. But SAZ poses some significant risks. It is highly toxic and can react to form potentially explosive compounds. Azide reagents and intermediates react with some metals5, strong acids, and certain chlorinated solvents6 and this needs to be considered when using SAZ or when developing routes that involve azide-containing intermediates. Health Hazards & Physical Hazards of SAZ Hazard Statements Fatal if swallowed or in contact with skin. Very toxic to aquatic life with long lasting effects. Health Hazards: SAZ is highly toxic when ingested orally or absorbed through the skin. Azides form strong complexes with hemoglobin, and consequently block oxygen transport in the blood. They are more harmful to the heart and the brain than to other organs, because the heart and the brain use a lot of oxygen. Symptoms of exposure include headache, dizziness, nausea and vomiting, rapid breathing and heart rate, and skin burns and blisters (direct skin contact) and in the case of serious overexposure, convulsions and death. It will also react with acids to form hydrazoic acid (HN3). Unlike sodium azide, which is a crystalline solid, hydrazoic acid is a low-boiling, volatile, liquid. Hydrazoic acid is also highly toxic and its volatility makes it more readily inhaled causing lung irritation and potentially bronchitis and lung edema. -

Aziridination of Alkenes Promoted by Iron Or Ruthenium Complexes
Aziridination of Alkenes Promoted by Iron or Ruthenium Complexes Caterina Damiano, Daniela Intrieri and Emma Gallo* Department of Chemistry, University of Milan, Via C. Golgi 19, 20133 Milan (Italy). E-mail address: [email protected]. Keywords: Aziridines, Nitrene reagents, Alkenes, Homogenous catalysis, Iron, Ruthenium. Abstract Molecules containing an aziridine functional group are a versatile class of organic synthons due to the presence of a strained three member, which can be easily involved in ring-opening reactions and the aziridine functionality often show interesting pharmaceutical and/or biological behaviours. For these reasons, the scientific community is constantly interested in developing efficient procedures to introduce an aziridine moiety into organic skeletons and the one-pot reaction of an alkene double bond with a nitrene [NR] source is a powerful synthetic strategy. Herein we describe the catalytic activity of iron or ruthenium complexes in promoting the reaction stated above by stressing the potential and limits of each synthetic protocol. 1. Introduction Aziridines, the smallest N-heterocycle compounds, have attracted considerable attention in the last few decades due to their many applications in biological and synthetic chemistry [1]. The aziridine functionality is often responsible for the activity of biologically active species (such as antitumor compounds, antibiotics and enzyme inhibitors) and aziridine containing molecules [2] are also useful building blocks in the synthesis of fine chemicals and pharmaceuticals [3-6]. The striking chemical properties of aziridines are due to the energy associated to the strained three- membered ring [7], which renders them very active and versatile starting materials for the synthesis of several useful molecules such as amines, amino acids, β-lactams, polymers and α-amido ketones [8, 9]. -

Alcohol Oxidation
Alcohol oxidation Alcohol oxidation is an important organic reaction. Primary alcohols (R-CH2-OH) can be oxidized either Mechanism of oxidation of primary alcohols to carboxylic acids via aldehydes and The indirect oxidation of aldehyde hydrates primary alcohols to carboxylic acids normally proceeds via the corresponding aldehyde, which is transformed via an aldehyde hydrate (R- CH(OH)2) by reaction with water. The oxidation of a primary alcohol at the aldehyde level is possible by performing the reaction in absence of water, so that no aldehyde hydrate can be formed. Contents Oxidation to aldehydes Oxidation to ketones Oxidation to carboxylic acids Diol oxidation References Oxidation to aldehydes Oxidation of alcohols to aldehydes is partial oxidation; aldehydes are further oxidized to carboxylic acids. Conditions required for making aldehydes are heat and distillation. In aldehyde formation, the temperature of the reaction should be kept above the boiling point of the aldehyde and below the boiling point of the alcohol. Reagents useful for the transformation of primary alcohols to aldehydes are normally also suitable for the oxidation of secondary alcohols to ketones. These include: Oxidation of alcohols to aldehydes and ketones Chromium-based reagents, such as Collins reagent (CrO3·Py2), PDC or PCC. Sulfonium species known as "activated DMSO" which can result from reaction of DMSO with electrophiles, such as oxalyl chloride (Swern oxidation), a carbodiimide (Pfitzner-Moffatt oxidation) or the complex SO3·Py (Parikh-Doering oxidation). Hypervalent iodine compounds, such as Dess-Martin periodinane or 2-Iodoxybenzoic acid. Catalytic TPAP in presence of excess of NMO (Ley oxidation). Catalytic TEMPO in presence of excess bleach (NaOCl) (Oxoammonium-catalyzed oxidation). -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Synthesis of Alkynyl Ribofuranosides
City University of New York (CUNY) CUNY Academic Works Dissertations and Theses City College of New York 2011 Synthesis of Alkynyl Ribofuranosides Christian Rodriguez CUNY City College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/cc_etds_theses/25 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] SYNTHESIS OF ALKYNYL RIBOFURANOSIDES A Thesis Presented to The Faculty of the Chemistry Program The City College of New York In (Partial) Fulfillment of the Requirements for the Degree Master of Arts by Christian Rodriguez December, 2010 - 1 - Synthesis of Alkynyl Ribofuranosides By Christian Rodriguez Mentor: P. Meleties Table of Contents Chapter 1 1.1 Introduction 6 1.2 Preparation of ribonolactone template 8 1.3 Synthesis of protected ribonolactone 9 Chapter 2 2.1 Preparation of ethynyl ribofuranosides 10 2.2 Reaction with ethynylmagnesium bromide 12 2.3 Intramolecular cyclization of diyne diol 14 2.4 Reaction of ribonolactone with lithium acetylide 15 2.5 Boron trifluoride hemiacetal deoxygenation 15 2.6 Alternative deacetylation 16 Chapter 3 3.1 Selecting appropriate protecting group 19 3.2 Preparation of trimethylsilyl alkynyl 5-O-benzyl-2,3-O isopropylidene 19 ribofuranoside 3.3 Lewis acid promoted triethylsilane dehydroxylation mechanism 20 Chapter 4 4.1 Hemiacetal alkynylation 25 4.2 Hemiacetal nucleophilic addition mechanism 26 4.3 Intramolecular -
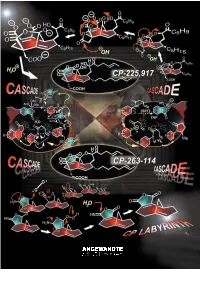
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

20 More About Oxidation–Reduction Reactions
More About 20 Oxidation–Reduction Reactions OOC n important group of organic reactions consists of those that O A involve the transfer of electrons C from one molecule to another. Organic chemists H OH use these reactions—called oxidation–reduction reactions or redox reactions—to synthesize a large O variety of compounds. Redox reactions are also important C in biological systems because many of these reactions produce HH energy. You have seen a number of oxidation and reduction reactions in other chapters, but discussing them as a group will give you the opportunity to CH3OH compare them. In an oxidation–reduction reaction, one compound loses electrons and one com- pound gains electrons. The compound that loses electrons is oxidized, and the one that gains electrons is reduced. One way to remember the difference between oxidation and reduction is with the phrase “LEO the lion says GER”: Loss of Electrons is Oxi- dation; Gain of Electrons is Reduction. The following is an example of an oxidation–reduction reaction involving inorganic reagents: Cu+ + Fe3+ ¡ Cu2+ + Fe2+ In this reaction,Cu+ loses an electron, so Cu+ is oxidized. Fe3+ gains an electron, so Fe3+ is reduced. The reaction demonstrates two important points about oxidation– reduction reactions. First, oxidation is always coupled with reduction. In other words, a compound cannot gain electrons (be reduced) unless another compound in the reaction simultaneously loses electrons (is oxidized). Second, the compound that is oxidized (Cu+) is called the reducing agent because it loses the electrons that are used to reduce the other compound (Fe3+). Similarly, the compound that is reduced (Fe3+) is called the oxidizing agent because it gains the electrons given up by the other compound (Cu+) when it is oxidized. -

Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2011 Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent Costyl Ngnouomeuchi Njiojob [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Heterocyclic Compounds Commons, Organic Chemicals Commons, and the Polycyclic Compounds Commons Recommended Citation Njiojob, Costyl Ngnouomeuchi, "Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent. " PhD diss., University of Tennessee, 2011. https://trace.tennessee.edu/utk_graddiss/1210 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Costyl Ngnouomeuchi Njiojob entitled "Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Chemistry. David C. Baker, Major Professor We have read this dissertation and recommend its acceptance: Shawn Campagna, Ben Xue, Elizabeth Howell Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Costyl Ngnouomeuchi Njiojob December 2011 DEDICATION This dissertation is dedicated to my Parents Francois Ngnouomeuchi and Lydie T. -

The Synthetic-Technical Development of Oseltamivir Phosphate Tamiflu<Sup>
HOT TOPICS: SANDMEIER PRIZE 2006 93 doi:10.2533/chimia.2007.93 CHIMIA 2007, 61, No. 3 Chimia 61 (2007) 93–99 © Schweizerische Chemische Gesellschaft ISSN 0009–4293 The Synthetic-Technical Development of Oseltamivir Phosphate TamifluTM: A Race against Time Stefan Abrechta , Muriel Cordon Federspielb , Heinrich Estermannb , Rolf Fischerb , Martin Karpf*a , Hans-Jürgen Mairb , Thomas Oberhauserc , Gösta Rimmlerb , René Trussardia , and Ulrich Zuttera Recipients of the Sandmeyer Prize 2006 of the Swiss Chemical Society Abstract: The clinical development of the first orally available neuraminidase inhibitor prodrug oseltamivir phos- phate (TamifluTM) proceeded very fast. In order to support this program an unprecedented team effort in chemical process research, development, piloting, production and analytics took place, which allowed the successful launch of TamifluTM in 1999, only two and a half years after it was licensed from Gilead Sciences. This article describes selected aspects of the commercially used synthesis route and a brief summary of alternative syntheses devised by Roche chemists. Keywords: Analytics · Influenza neuraminidase inhibitor · Oseltamivir phosphate · Production · Synthesis · Technical development 1. Introduction H O H O CO H In September 1996 a license agreement be- H O 2 O CO Et tween Gilead Sciences, Foster City, Califor- 2 H O nia and F. Hoffmann-La Roche Ltd, Basel AcHN H N NH was signed for the co-development of the AcHN 2 NH2 •H3 PO4 novel orally available neuraminidase inhibi- NH tor prodrug molecule 1 (Fig. 1) patented by Gilead Sciences in February 1995,[1] today 1 2 known as oseltamivir phosphate, trade name TamifluTM. In April 1999, after only two and Oseltamivir phosphate Zanamivir a half years of development time, a new drug TamifluTM RelenzaTM GS-4104-02 GG-167 application (NDA) for 1 was filed with the RO0640796-002 US Food and Drug Administration (FDA) for the use of 1 for the treatment of influenza Fig.