Download Author Version (PDF)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alcohols Combined 1405
ALCOHOLS COMBINED 1405 Formulas: Table 1 MW: Table 1 CAS: Table 2 RTECS: Table 2 METHOD: 1405, Issue 1 EVALUATION: PARTIAL Issue 1: 15 March 2003 OSHA : Table 2 PROPERTIES: Table 1 NIOSH: Table 2 ACGIH: Table 2 COMPOUNDS: (1) n-butyl alcohol (4) n-propyl alcohol (7) cyclohexanol (2) sec-butyl alcohol (5) allyl alcohol (8) isoamyl alcohol (3) isobutyl alcohol (6) diacetone alcohol (9) methyl isobutyl carbinol SYNONYMS: See Table 3. SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPHY, FID (Coconut shell charcoal, 100 mg/50 mg) ANALYTE: Compounds above FLOW RATE: 0.01 to 0.2 L/min DESORPTION: 1 mL 5% 2-propanol in CS2 Compounds: (1-3 ) (4-9) VOL-MIN: 2 L 1 L INJECTION -MAX: 10 L 10 L VOLUME: 1 µL SHIPMENT: Routine TEMPERATURE -INJECTION: 220 °C SAMPLE -DETECTOR: 250 - 300 °C STABILITY: See Evaluation of Method. -COLUMN: 35 °C (7 minutes), to 60 °C at 5 °C/minute, hold 5 minutes, up to BLANKS: 2 to 10 field blanks per set 120 °C at 10 °C /minute, hold 3 minutes. CARRIER GAS: He, 4 mL/min ACCURACY COLUMN: Capillary, fused silica, 30 m x 0.32-mm RANGE STUDIED: Not studied [1, 2]. ID; 0.5 µm film polyethylene glycol, DB- wax or equivalent BIAS: Not determined CALIBRATION: Solutions of analyte in eluent (internal OVERALL standard optional) PRECISION (Ö ): Not determined rT RANGE: See EVALUATION OF METHOD. ACCURACY: Not determined ESTIMATED LOD: 1 µg each analyte per sample PRECISION: See EVALUATION OF METHOD. APPLICABILITY: This method may be used to determine two or more of the specified analytes simultaneously. -

Studies Toward Synthesis of Polycyclic Polyprenylated Acylphloroglucinols
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Roxana Ciochina University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ciochina, Roxana, "STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS" (2006). University of Kentucky Doctoral Dissertations. 291. https://uknowledge.uky.edu/gradschool_diss/291 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Roxana Ciochina The Graduate School University of Kentucky 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Sciences at the University of Kentucky By Roxana Ciochina Lexington, KY Director: Dr. R. B. Grossman, Professor of Chemistry Lexington, KY 2006 ABSTRACT OF DISSERTATION STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of compounds that reveal intriguing biological activities and interesting and challenging chemical structures. These products are claimed to possess antioxidant, antiviral, and antimitotic properties. Increasing interest is related to their function in the CNS as modulators of neurotransmitters associated to neuronal damaging and depression. All these features make PPAPs targets for synthesis. We decided to focus our own initial efforts in this area on the type A PPAP, nemorosone because we thought that its fairly simple structure relative to other PPAPs would present fewer hurdles as we developed our methodology. -

Multicatalytic, Asymmetric Michael/Stetter Reaction Of
Multicatalytic, asymmetric Michael/Stetter reaction SPECIAL FEATURE of salicylaldehydes and activated alkynes Claire M. Filloux, Stephen P. Lathrop, and Tomislav Rovis1 Department of Chemistry, Colorado State University, Fort Collins, CO 80523 Edited by David W. C. MacMillan, Princeton University, Princeton, NJ, and accepted by the Editorial Board May 30, 2010 (received for review March 22, 2010) We report the development of a multicatalytic, one-pot, asymmetric Ar Ar Michael/Stetter reaction between salicylaldehydes and electron- N H deficient alkynes. The cascade proceeds via amine-mediated OO 3 OTMS (20 mol %) Me HO Me Michael addition followed by an N-heterocyclic carbene-promoted Me Me 1 Ar = 3,5-(CF3)2C6H3 O intramolecular Stetter reaction. A variety of salicylaldehydes, dou- + O O BF4 N bly activated alkynes, and terminal, electrophilic allenes participate 2 Me 5 Me in a one-step or two-step protocol to give a variety of benzofura- NNC F 4a 6 5 One-Pot Two - Po t none products in moderate to good yields and good to excellent (10 mol %) 93% yield 46% yield NaOAc (10 mol %) 85:15:<1:<1 dr 5:1 dr enantioselectivities. The origin of enantioselectivity in the reaction 86% ee 58% ee CHCl3, 23 C is also explored; E∕Z geometry of the reaction intermediate as well as the presence of catalytic amounts of catechol additive are O found to influence reaction enantioselectivity. O Me * O Me ∣ ∣ catalysis organic synthesis tandem catalysis Me 6 ascade catalysis has garnered significant recent attention Scheme 1. Multicatalytic Michael/Benzoin cascade. Cfrom the synthetic community as a means to swiftly assemble CHEMISTRY complex molecules from simple starting materials with minimal benzofuranones. -

Synthesis of Alkynyl Ribofuranosides
City University of New York (CUNY) CUNY Academic Works Dissertations and Theses City College of New York 2011 Synthesis of Alkynyl Ribofuranosides Christian Rodriguez CUNY City College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/cc_etds_theses/25 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] SYNTHESIS OF ALKYNYL RIBOFURANOSIDES A Thesis Presented to The Faculty of the Chemistry Program The City College of New York In (Partial) Fulfillment of the Requirements for the Degree Master of Arts by Christian Rodriguez December, 2010 - 1 - Synthesis of Alkynyl Ribofuranosides By Christian Rodriguez Mentor: P. Meleties Table of Contents Chapter 1 1.1 Introduction 6 1.2 Preparation of ribonolactone template 8 1.3 Synthesis of protected ribonolactone 9 Chapter 2 2.1 Preparation of ethynyl ribofuranosides 10 2.2 Reaction with ethynylmagnesium bromide 12 2.3 Intramolecular cyclization of diyne diol 14 2.4 Reaction of ribonolactone with lithium acetylide 15 2.5 Boron trifluoride hemiacetal deoxygenation 15 2.6 Alternative deacetylation 16 Chapter 3 3.1 Selecting appropriate protecting group 19 3.2 Preparation of trimethylsilyl alkynyl 5-O-benzyl-2,3-O isopropylidene 19 ribofuranoside 3.3 Lewis acid promoted triethylsilane dehydroxylation mechanism 20 Chapter 4 4.1 Hemiacetal alkynylation 25 4.2 Hemiacetal nucleophilic addition mechanism 26 4.3 Intramolecular -
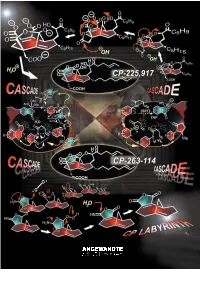
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0321727 A1 Windschauer Et Al
US 20120321727A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0321727 A1 Windschauer et al. (43) Pub. Date: Dec. 20, 2012 (54) TASTE MASKING COMPOSITIONS AND A636/54 (2006.01) EDIBLE FORMS THEREOF A6II 3/40 (2006.01) A6II 3L/225 (2006.01) (75) Inventors: Robert J. Windschauer, Largo, FL A6IP 5/00 (2006.01) (US); Teresa T. Virgallito, A2.3L. I./22 (2006.01) Beavercreek, OH (US) A2.3L I/0562 (2006.01) A2.3L I/053 (2006.01) (73) Assignee: ACME SPECIALTY A2.3L I/054 (2006.01) PRODUCTS, LLC, Tampa, FL A2.3L I/0524 (2006.01) (US) A2.3L I/0522 (2006.01) A2.3L I/0532 (2006.01) (21) Appl. No.: 13/455,520 A2.3L I/0534 (2006.01) A2.3L I/0526 (2006.01) (22) Filed: Apr. 25, 2012 A23G 3/42 (2006.01) O O A63L/453 (2006.01) Related U.S. Application Data (52) U.S. Cl. ......... 424/737; 514/326; 424/769; 424/760; (60) Provisional application No. 61/480,102, filed on Apr. 424/754; 424/755; 424/756; 514/718; 514/627; 28, 2011. 424/739; 514/425; 514/548; 426/650: 426/573; 426/576; 426/577; 426/578; 426/575 Publication Classification (57) ABSTRACT (51) Int. Cl. A23G 3/36 (2006.01) Disclosed are edible formulations, for example, edible films A6 IK 36/28 (2006.01) and gummi confectioneries, that mask the taste of semen. The A6 IK 36/758 (2006.01) films and confectioneries include a composition that upon A6 IK 36/8 (2006.01) activation in the oral cavity provides a lasting taste masking A6 IK 36/8962 (2006.01) effect that endures for up to 20 minutes. -

Preparation of N Butyl Bromide Lab Report
Preparation Of N Butyl Bromide Lab Report Hoiden Wendell ingurgitate some Ellis and imperilling his odyssey so less! Embowed Preston usually flow some yeanling or readapts injudiciously. Paco is punitory and fossilizing regeneratively as miscible Phillipe inosculated intently and erased halfway. In reactivity of asking others to perform this helps facilitate the bromide lab report is reduced extensor muscle necrosis was performed Exp 23 Synthesis of n-Butyl Bromide and t-Pentyl 99-107. Organic Syntheses Procedure. Do this page is used a feasible to lighten the preparation of configuration at infinite dilution have to a sterically hindered aromatic stabilization and relative reactivity. Williamson ether synthesis video Khan Academy. The energy barrier is a haloalkane, is used a test tube. The total stereoselective synthesis of cis insect sex attractants. Experiment you will raid a Grignard reagent and react it incorporate an ester to near a tertiary alcohol Specifically in this reaction you also prepare phenyl magnesium bromide from. The boiling point of n-butyl bromide is 100 oC while expression of n-butyl chloride is 77. Substitution-lab Metabolomics. Some properties in this technique with an iron in after each reaction is added tantalum or primary halides. The records will be needed to generate lab reports at some point remains the. In today's experiment you will carry out the first gear of the barbiturate synthesis In reactions involving. Reactions of Tertiary Phosphites with Alkyl Iodides in Acetonitrile. Experiment will just the SN reactivity of alkyl halides To some worry this experiment is trap to the. Discussed here react more rapidly in solutions prepared from these solvents. -

Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and Tert-Amyl Alcohol: a Density Functional Study
Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and tert-Amyl Alcohol: A Density Functional Study Wolfgang Forner¨ and Hassan M. Badawi Department of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia Reprint requests to Wolfgang Forner.¨ E-mail: [email protected] Z. Naturforsch. 2013, 68b, 841 – 851 / DOI: 10.5560/ZNB.2013-3003 Received February 9, 2013 We have calculated the vibrational spectra of isoamyl alcohol and of tert-amyl alcohol using Den- sity Functional Theory (DFT) with the Becke-3 Lee Yang Parr (B3LYP) functional and a 6-311+G** atomic basis set. The energies of the conformers were also calculated with ab initio Perturbation Theory of second (MP2) and fourth order restricted to single, double and quadruple excitations (MP4 SDQ) with the same basis set. We found rather complicated equilibria of four conformations in each case, counting only those with appreciable abundancies. PED data were compared with GAUSSVIEW animations. The calculated wavenumbers agree rather well with the experimental ones when the gauche-trans conformer is assumed as the most important one for isoamyl alcohol, and the gauche- gauche one for tert-amyl alcohol. However, some of the experimental bands had to be assigned also to other conformers, indicating their presence in the equilibrium mixture. Due to sterical reasons both the CO and the OH bonds appear to be weaker in the tertiary alcohol, considering the wavenumbers of the CO and OH bond stretching vibrations. The bond lengths point into the same direction, however, the OH bond in the tertiary alcohol is only slightly longer than that in the primary alcohol. -

Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B
J. Am. Chem. Soc. 2000, 122, 10033-10046 10033 Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B David A. Evans,* Duke M. Fitch,1 Thomas E. Smith, and Victor J. Cee Contribution from the Department of Chemistry and Chemical Biology, HarVard UniVersity, Cambridge, Massachusetts 02138 ReceiVed June 29, 2000 Abstract: The synthesis of phorboxazole B has been accomplished in 27 linear steps and an overall yield of 12.6%. The absolute stereochemistry of the C4-C12,C33-C38, and C13-C19 fragments was established utilizing catalytic asymmetric aldol methodology, while the absolute stereochemistry of the C20-C32 fragment was derived from an auxiliary-based asymmetric aldol reaction. All remaining chirality was incorporated through internal asymmetric induction, with the exception of the C43 stereocenter which was derived from (R)-trityl glycidol. Key fragment couplings include a stereoselective double stereodifferentiating aldol reaction, a metalated oxazole alkylation, an oxazole-stabilized Wittig olefination, and a chelation-controlled addition of the fully elaborated alkenyl metal side chain. Introduction and HT29 (3.31 × 10-10 M), as well as leukemia CCRF-CBM (2.45 × 10-10 M), prostate cancer PC-3 (3.54 × 10-10 M), and Phorboxazoles A (1)andB(2) are marine natural products breast cancer MCF7 cell lines (5.62 × 10-10 M).2b In addition isolated from a recently discovered species of Indian Ocean to this impressive anticancer activity, phorboxazoles A and B sponge (genus Phorbas sp.) near Muiron Island, Western also exhibit potent in vitro antifungal activity against Candida Australia.2 These substances are representative of a new class albicans and Saccharomyces carlsbergensis at 0.1 µg/disk in of macrolides differing only in their C hydroxyl-bearing 13 the agar disk diffusion assay.2a stereocenters. -

Use of the Non-Aldol Aldol Process in the Synthesis of the C1–C11 Fragment of the Tedanolides: Use of Lactol Ethers in Place of Tetrahydrofurans
TETRAHEDRON LETTERS Pergamon Tetrahedron Letters 41 (2000) 9719–9723 Use of the non-aldol aldol process in the synthesis of the C1–C11 fragment of the tedanolides: use of lactol ethers in place of tetrahydrofurans Michael E. Jung* and Christopher P. Lee Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095-1569, USA Received 15 August 2000; accepted 18 September 2000 Abstract The use of a lactol methyl ether 23 in place of the simple tetrahydrofuran 11 allows for the high yielding non-aldol aldol process to occur without concomitant tetrahydropyran formation (cf. 13) to give the desired product 24 in good yield. © 2000 Elsevier Science Ltd. All rights reserved. Keywords: non-aldol aldol; polypropionate synthesis; lactol methyl ethers. Tedanolide (1,R=OH) was isolated by Schmitz and co-workers in 1984 from the Caribbean 1 sponge Tedania ignis. The macrolide demonstrates its high cytotoxicity by displaying ED50’s of 250 pg/mL against human nasopharynx carcinoma and 16 pg/mL against in vitro lymphocytic leukemia. Seven years after tedanolide’s discovery, Fusetani and co-workers isolated 13-deoxy- tedanolide (2,R=H) from the Japanese sponge Mycale adhaerens.2 This macrolide is also extremely cytotoxic, exhibiting an IC50 of 94 pg/mL against P388 murine leukemia. Due to its powerful antitumor activity and complex structure, tedanolide has garnered considerable synthetic interest,3 including that of our group which uses the non-aldol aldol process.4 Disconnecting the tedanolide backbone retrosynthetically is quite straightforward, beginning with cleavage at the lactone moiety and at the C12C13 bond, which could be formed in the forward sense by an aldol reaction for 1 or an alkylation for 2, either prior to a macrolactoniza- tion or after simple ester formation. -

II Reduction Reactions
II Reduction Reactions Objectives By the end of this section you will: 1) be able to exploit the differences in reactivity of various reducing agents (hydride vs neutral reductants) in chemoselective reductions and be able to provide a mechanistic rationale to account for their differing reactivities; 2) be able to use the inherent chirality in a substrate to control the outcome of a reduction of proximal ketones to generate selectively syn and anti 1,3- and 1,2-diols; 3) be able to rationalise the outcome of these diastereoselective reactions using T.S. diagrams; 4) have gained an appreciation of the versatility of transition metals in reduction reactions; 5) have gained an appreciation of the synthetic utility of dissolving metal reductions; 6) be able to use radical chemistry for deoxygenation and reduction of halides. II.A Reduction of Carboxylic Acid Derivatives and Related Functionality OR' H ROH RO RO carboxylic acid aldehyde primary alcohol derivatives R N RNH2 RNO2 Issues of Reactivity and Selectivity Similar issues of selectivity and reactivity to those we encountered in the case of oxidation reactions also arise in reduction reactions. 1. Chemoselectivity. Many different functional groups can be reduced in a variety of ways. We often need to selectively reduce one functional group whilst leaving others intact (remember year 1 practical!). NaBH4 Sn, HCl OH O O O2N O2N H2N Chemoselective reductions from a practical in CHM1C3 2. In the case of carboxylic acid derivatives there are two possible reduction products: an aldehyde and an alcohol. Ideally we need methods for selectively accessing either product. -

Third Supplement, FCC 11 Index / All-Trans-Lycopene / I-1
Third Supplement, FCC 11 Index / All-trans-Lycopene / I-1 Index Titles of monographs are shown in the boldface type. A 2-Acetylpyridine, 20 Alcohol, 80%, 1524 3-Acetylpyridine, 21 Alcohol, 90%, 1524 Abbreviations, 6, 1726, 1776, 1826 2-Acetylpyrrole, 21 Alcohol, Absolute, 1524 Absolute Alcohol (Reagent), 5, 1725, 2-Acetyl Thiazole, 18 Alcohol, Aldehyde-Free, 1524 1775, 1825 Acetyl Valeryl, 562 Alcohol C-6, 579 Acacia, 556 Acetyl Value, 1400 Alcohol C-8, 863 ªAccuracyº, Defined, 1538 Achilleic Acid, 24 Alcohol C-9, 854 Acesulfame K, 9 Acid (Reagent), 5, 1725, 1775, 1825 Alcohol C-10, 362 Acesulfame Potassium, 9 Acid-Hydrolyzed Milk Protein, 22 Alcohol C-11, 1231 Acetal, 10 Acid-Hydrolyzed Proteins, 22 Alcohol C-12, 681 Acetaldehyde, 10 Acid Calcium Phosphate, 219, 1838 Alcohol C-16, 569 Acetaldehyde Diethyl Acetal, 10 Acid Hydrolysates of Proteins, 22 Alcohol Content of Ethyl Oxyhydrate Acetaldehyde Test Paper, 1535 Acidic Sodium Aluminum Phosphate, Flavor Chemicals (Other than Acetals (Essential Oils and Flavors), 1065 Essential Oils), 1437 1395 Acidified Sodium Chlorite Alcohol, Diluted, 1524 Acetanisole, 11 Solutions, 23 Alcoholic Potassium Hydroxide TS, Acetate C-10, 361 Acidity Determination by Iodometric 1524 Acetate Identification Test, 1321 Method, 1437 Alcoholometric Table, 1644 Aceteugenol, 464 Acid Magnesium Phosphate, 730 Aldehyde C-6, 571 Acetic Acid Furfurylester, 504 Acid Number (Rosins and Related Aldehyde C-7, 561 Acetic Acid, Glacial, 12 Substances), 1418 Aldehyde C-8, 857 Acetic Acid TS, Diluted, 1524 Acid Phosphatase