Control of Innate Immunity by Sialic Acids in the Nervous Tissue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vocabulario De Morfoloxía, Anatomía E Citoloxía Veterinaria
Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) Servizo de Normalización Lingüística Universidade de Santiago de Compostela COLECCIÓN VOCABULARIOS TEMÁTICOS N.º 4 SERVIZO DE NORMALIZACIÓN LINGÜÍSTICA Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) 2008 UNIVERSIDADE DE SANTIAGO DE COMPOSTELA VOCABULARIO de morfoloxía, anatomía e citoloxía veterinaria : (galego-español- inglés) / coordinador Xusto A. Rodríguez Río, Servizo de Normalización Lingüística ; autores Matilde Lombardero Fernández ... [et al.]. – Santiago de Compostela : Universidade de Santiago de Compostela, Servizo de Publicacións e Intercambio Científico, 2008. – 369 p. ; 21 cm. – (Vocabularios temáticos ; 4). - D.L. C 2458-2008. – ISBN 978-84-9887-018-3 1.Medicina �������������������������������������������������������������������������veterinaria-Diccionarios�������������������������������������������������. 2.Galego (Lingua)-Glosarios, vocabularios, etc. políglotas. I.Lombardero Fernández, Matilde. II.Rodríguez Rio, Xusto A. coord. III. Universidade de Santiago de Compostela. Servizo de Normalización Lingüística, coord. IV.Universidade de Santiago de Compostela. Servizo de Publicacións e Intercambio Científico, ed. V.Serie. 591.4(038)=699=60=20 Coordinador Xusto A. Rodríguez Río (Área de Terminoloxía. Servizo de Normalización Lingüística. Universidade de Santiago de Compostela) Autoras/res Matilde Lombardero Fernández (doutora en Veterinaria e profesora do Departamento de Anatomía e Produción Animal. -

Cell Structure and Function
BIOLOGY Chapter 4: pp. 59-84 10th Edition Cell Structure and S. Sylvia Function Plasma membrane: outer surface that Ribosome: Fimbriae: regulates entrance site of protein synthesis hairlike bristles that and exit of molecules allow adhesion to Mader the surfaces Inclusion body: Conjugation pilus: stored nutrients for elongated, hollow later use appendage used for DNA transfer to other Nucleus: Mesosome: bacterial cells plasma membrane Cytoskeleton: maintains cell that folds into the Nucleoid: shape and assists cytoplasm and location of the bacterial movement of increases surface area chromosome cell parts: Endoplasmic Plasma membrane: reticulum: sheath around cytoplasm that regulates entrance and exit of molecules Cell wall: covering that supports, shapes, and protects cell Glycocalyx: gel-like coating outside cell wall; if compact, called a capsule; if diffuse, called a slime layer Flagellum: rotating filament present in some bacteria that pushes the cell forward *not in plant cells PowerPoint® Lecture Slides are prepared by Dr. Isaac Barjis, Biology Instructor 1 Copyright © The McGraw Hill Companies Inc. Permission required for reproduction or display Outline Cellular Level of Organization Cell theory Cell size Prokaryotic Cells Eukaryotic Cells Organelles Nucleus and Ribosome Endomembrane System Other Vesicles and Vacuoles Energy related organelles Cytoskeleton Centrioles, Cilia, and Flagella 2 Cell Theory Detailed study of the cell began in the 1830s A unifying concept in biology Originated from the work of biologists Schleiden and Schwann in 1838-9 States that: All organisms are composed of cells German botanist Matthais Schleiden in 1838 German zoologist Theodor Schwann in 1839 All cells come only from preexisting cells German physician Rudolph Virchow in 1850’s Cells are the smallest structural and functional unit of organisms 3 Organisms and Cells Copyright © The McGraw-Hill Companies, Inc. -

NIH Public Access Author Manuscript Mucosal Immunol
NIH Public Access Author Manuscript Mucosal Immunol. Author manuscript; available in PMC 2014 March 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mucosal Immunol. 2013 March ; 6(2): 379–392. doi:10.1038/mi.2012.81. Molecular Organization of the Mucins and Glycocalyx Underlying Mucus Transport Over Mucosal Surfaces of the Airways Mehmet Kesimer*,‡,▫, Camille Ehre*, Kimberly A. Burns*, C. William Davis*,†, John K. Sheehan*,‡, and Raymond J. Pickles*,§ *Cystic Fibrosis/Pulmonary Research and Treatment Center, University of North Carolina, Chapel Hill, NC 27599-7248 ‡Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC 27599-7248 †Department of Cell and Molecular Physiology, University of North Carolina, Chapel Hill, NC 27599-7248 §Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC 27599-7248 Abstract Mucus, with its burden of inspired particulates, and pathogens, is cleared from mucosal surfaces of the airways by cilia beating within the periciliary layer (PCL). The PCL is held to be ‘watery’ and free of mucus by thixotropic-like forces arising from beating cilia. With radii of gyration ~250 nm, however, polymeric mucins should reptate readily into the PCL, so we assessed the glycocalyx for barrier functions. The PCL stained negative for MUC5AC and MUC5B, but it was positive for keratan sulfate, a glycosaminoglycan commonly associated with glycoconjugates. Shotgun proteomics showed keratan sulfate-rich fractions from mucus containing abundant tethered mucins, MUC1, MUC4, and MUC16, but no proteoglycans. Immuno-histology by light and electron microscopy localized MUC1 to microvilli, MUC4 and MUC20 to cilia, and MUC16 to goblet cells. -

Nanoarchitecture and Dynamics of the Mouse Enteric Glycocalyx Examined by Freeze-Etching Electron Tomography and Intravital Microscopy
ARTICLE https://doi.org/10.1038/s42003-019-0735-5 OPEN Nanoarchitecture and dynamics of the mouse enteric glycocalyx examined by freeze-etching electron tomography and intravital microscopy Willy W. Sun1,2,5, Evan S. Krystofiak1,5, Alejandra Leo-Macias1, Runjia Cui1, Antonio Sesso3, Roberto Weigert 4, 1234567890():,; Seham Ebrahim4 & Bechara Kachar 1* The glycocalyx is a highly hydrated, glycoprotein-rich coat shrouding many eukaryotic and prokaryotic cells. The intestinal epithelial glycocalyx, comprising glycosylated transmembrane mucins, is part of the primary host-microbe interface and is essential for nutrient absorption. Its disruption has been implicated in numerous gastrointestinal diseases. Yet, due to chal- lenges in preserving and visualizing its native organization, glycocalyx structure-function relationships remain unclear. Here, we characterize the nanoarchitecture of the murine enteric glycocalyx using freeze-etching and electron tomography. Micrometer-long mucin filaments emerge from microvillar-tips and, through zigzagged lateral interactions form a three-dimensional columnar network with a 30 nm mesh. Filament-termini converge into globular structures ~30 nm apart that are liquid-crystalline packed within a single plane. Finally, we assess glycocalyx deformability and porosity using intravital microscopy. We argue that the columnar network architecture and the liquid-crystalline packing of the fila- ment termini allow the glycocalyx to function as a deformable size-exclusion filter of luminal contents. 1 Laboratory of Cell Structure and Dynamics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD 20892, USA. 2 Neuroscience and Cognitive Science Program, University of Maryland, College Park, MD 20740, USA. 3 Sector of Structural Biology, Institute of Tropical Medicine, University of São Paulo, Sao Paulo, SP 05403, Brazil. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Mechanical Characterisation of Bone Cells and Their Glycocalyx
Mechanical characterisation of bone cells and their glycocalyx a dissertation presented by Stefania Marcotti to The Department of Mechanical Engineering University of Sheffield Sheffield, United Kingdom in partial fulfilment of the requirements for the degree of Doctor of Philosophy Supervisor: Prof. Damien Lacroix Co-Supervisor: Dr. Gwendolen C Reilly November 2017 Abstract Mechanotransductionreferstotheprocessbywhichacellisabletotranslatemech- anical stimulation into biochemical signals. In bone, mechanotransduction regu- lates how cells detect environmental stimuli and use these to direct towards bone deposition or resorption. The mechanical properties of bone cells have an impact on the way mechanical stimulation is sensed, however, little evidence is available about how these properties influence mechanotransduction. The aim of the present Thesis was to quantify the mechanical properties of bone cells with a combined experimental and computational approach. Atomic force microscopy was employed to quantify the stiffness of bone cells and their gly- cocalyx. Changes in cell stiffness during osteocytogenesis were explored. Single molecule force spectroscopy of glycocalyx components was performed to evalu- ate their anchoring to the cytoskeleton. A single cell finite element model was designed to discern the contributions of sub-cellular components in response to simulated cell nano-indentation. Wide ranges of variation were found for bone cell stiffness and a method was proposed to determine suitable sample sizes to capture population heterogeneity. Bytargetingsinglecomponentsoftheboneglycocalyx, itwaspossibletohypothes- ise different mechanotransduction mechanisms depending on the hyaluronic acid attachment to the cytoskeleton. The developed computational framework showed similar results to the nano-indentation experiments and highlighted the role of the actin cytoskeleton in withstanding compression and distributing strain within the cell. iii Publications and conference presentations Book chapters Marcotti S and Reilly GC (2017). -

Immuno-Electron and Confocal Laser Scanning Microscopy of the Glycocalyx
biology Article Immuno-Electron and Confocal Laser Scanning Microscopy of the Glycocalyx Shailey Gale Twamley 1,2, Anke Stach 1, Heike Heilmann 3, Berit Söhl-Kielczynski 4, Verena Stangl 1,2, Antje Ludwig 1,2,*,† and Agnieszka Münster-Wandowski 3,*,† 1 Medizinische Klinik für Kardiologie und Angiologie, Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, 10117 Berlin, Germany; [email protected] (S.G.T.); [email protected] (A.S.); [email protected] (V.S.) 2 DZHK (German Centre for Cardiovascular Research), Partner Site, 10117 Berlin, Germany 3 Institute of Integrative Neuroanatomy, Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, 10117 Berlin, Germany; [email protected] 4 Institute for Integrative Neurophysiology—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, 10117 Berlin, Germany; [email protected] * Correspondence: [email protected] (A.L.); [email protected] (A.M.-W.) † These authors equally contributed. Simple Summary: The glycocalyx (GCX) is a hydrated, gel-like layer of biological macromolecules attached to the cell membrane. The GCX acts as a barrier and regulates the entry of external substances into the cell. The function of the GCX is highly dependent on its structure and composition. Citation: Twamley, S.G.; Stach, A.; Pathogenic factors can affect the protective structure of the GCX. We know very little about the three- Heilmann, H.; Söhl-Kielczynski, B.; dimensional organization of the GXC. The tiny and delicate structures of the GCX are difficult Stangl, V.; Ludwig, A.; to study by microscopic techniques. -

Characteristics of a Glycoprotein in the Ocular Surface Glycocalyx
Investigative Ophthalmology & Visual Science, Vol. 33, No. 1, January 1992 Copyright © Association for Research in Vision and Ophthalmology Characteristics of a Glycoprotein in the Ocular Surface Glycocalyx llene K. Gipson,*t Michelle Yankauckas,* Sandra J. Spurr-Michaud,* Ann S. Tisdale,* and William Rineharr* A monoclonal antibody has been produced that binds to the apical squames (flattened cells) of the rat ocular surface epithelium and to the goblet cells of the conjunctiva. Immunoelectron microscopic localization of the antigen indicates that in apical cells it is present along the apical-microplical mem- brane in the region of the glycocalyx. In subapical squames, the antigen is in cytoplasmic vesicles. In some goblet cells, the antigen is in the Golgi network, and in others, it is located primarily in the membrane of the mucous granules. SDS-PAGE and immunoblot analysis demonstrate that the molecu- lar weight of the antigen is greater than 205 kD, and the electrophoretic band stains with Alcian blue followed by silver stain. Periodate oxidation of immunoblots and cryostat sections removes antibody binding. Neuraminidase treatment of cryostat sections does not remove antibody binding, whereas N-glycanase does. Taken together, these data indicate that the antigen recognized by the monoclonal antibody is a carbohydrate epitope on a high-molecular-weight, highly glycosylated glycoprotein in the glycocalyx of the ocular surface epithelium and goblet cell mucin granule membrane. The antigen appears to be stored within cytoplasmic vesicles and reaches the glycocalyx when cells differentiate to the apical-most position where the glycocalyx interfaces with the mucin layer of the tear film. Invest Ophthalmol Vis Sci 33:218-227,1992 The ocular surface epithelium that covers the con- tissue stained with tannic acid demonstrates that the junctiva and cornea is nonkeratinizing, stratified, and glycocalyx is a fine, filamentous layer. -

Biol 1020: a Tour of the Cell
Chapter 6: A Tour of the Cell Cell theory Cell organization and homeostasis Studying cells – microscopy and fractionation Eukaryotic vs. prokaryotic cells Compartments in eukaryotic cells (cell regions, organelles) Cytoskeleton Outside the cell . Chapter 6: A Tour of the Cell Cell theory Cell organization and homeostasis Studying cells – microscopy and fractionation Eukaryotic vs. prokaryotic cells Compartments in eukaryotic cells (cell regions, organelles) Cytoskeleton Outside the cell . • What are the main tenets of cell theory? • What are the major lines of evidence that all presently living cells have a common origin? . Cell theory All living organisms are composed of cells smallest “building blocks” of all multicellular organisms all cells are enclosed by a surface membrane that separates them from other cells and from their environment specialized structures with the cell are called organelles; many are membrane-bound . Cell theory Today, all new cells arise from existing cells All presently living cells have a common origin all cells have basic structural and molecular similarities all cells share similar energy conversion reactions all cells maintain and transfer genetic information in DNA the genetic code is essentially universal . • What are the main tenets of cell theory? • What are the major lines of evidence that all presently living cells have a common origin? . Chapter 6: A Tour of the Cell Cell theory Cell organization and homeostasis Studying cells – microscopy and fractionation Eukaryotic vs. prokaryotic cells Compartments in eukaryotic cells (cell regions, organelles) Cytoskeleton Outside the cell . • What is surface area to volume ratio, and why is it an important consideration for cells? • What (usually) happens to surface area to volume ratio as cells grow larger? . -

Introduction to Microbiology
Introduction to microbiology Prof. dr hab. Beata M. Sobieszczańska Department of Microbiology University of Medicine • http://www.lekarski.umed.wroc.pl/mikrobiologia • schedules, rules, important information • Consulting hours – teachers are always available for students during consulting hours or classes – apart from consulting hours – you must chase ! • Sick leaves (original) must be shown to the teacher just after an absence but not longer than after two weeks otherwise a sick note will not be honored - a copy of the sick note must be delivered to the secretary office • Class tests – 10 open questions • Terms: 1st, 2nd – if failed commission test from the whole material at the end of semester • Students with the average 4.8 will be released from the final exam • Presence on lectures and classes are obligatory • The final grade from classes is the average of all grades during semester Your best friend in this year: Medical Microbiology by Patrick R. Murray, Ken S. Rosenthal, Michael A. Pfaller Answer questions: • Name important cell wall structures of GP and GN bacteria • What is a role of these structures in human diseases? • Name other than bacterial cell wall structures and explain their role in bacterial pathogenicity • Do you understand the term pathogenicity? • Name five different genera GP and GN bacteria and indicate the colour they have after Gram staining Answer questions: • Name clinically important bacteria producing endospores – why endospores are important? • What is the difference between capsule and glycocalyx layer on GP bacteria? • What is axial filament? What role it plays? What bacteria produce axial filaments? • Name two types of pili and their role in bacterial pathogenicity Most bacteria come in one of three basic shapes: coccus, rod or bacillus & spiral MURRAY 7th ed. -

Defining the Layers of a Sensory Cilium with STORM and Cryo-Electron Nanoscopies
bioRxiv preprint doi: https://doi.org/10.1101/198655; this version posted October 6, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Defining the Layers of a Sensory Cilium with STORM and Cryo-Electron Nanoscopies. Authors: Michael A. Robichaux1, Valencia L. Potter1,2,3, Zhixian Zhang1, Feng He1, Michael F. Schmid1,4, Theodore G. Wensel1* 1Verna and Marrs McLean Department of Biochemistry and Molecular Biology; 2Graduate Program in Developmental Biology; 3Medical Scientist Training Program, Baylor College of Medicine, Houston, TX, 77030; 4Current address: SLAC Linear Accelerator Laboratory, Menlo Park, California 94025. *Correspondence: [email protected] Keywords: Connecting cilium, rod photoreceptor, STORM, super resolution, Bardet- Biedl syndrome 1 bioRxiv preprint doi: https://doi.org/10.1101/198655; this version posted October 6, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Primary cilia are cylindrical organelles extending from the surface of most animal cells that have been implicated in a host of signaling and sensory functions. Genetic defects in their component molecules, known as “ciliopathies” give rise to devastating symptoms, ranging from defective development, to kidney disease, to progressive blindness. The detailed structures of these organelles and the true functions of proteins encoded by ciliopathy genes are poorly understood because of the small size of cilia and the limitations of conventional microscopic techniques. We describe the combination of cryo-electron tomography, enhanced by sub-tomogram averaging, with super-resolution stochastic reconstruction microscopy (STORM) to define substructures and subdomains within the light-sensing rod sensory cilium of the mammalian retina. -
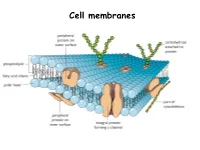
Applied Science Cell Membrane
Cell membranes Plasma Membrane Structure and Function The plasma membrane separates the internal environment of the cell from its surroundings. The plasma membrane is a phospholipid bilayer with embedded proteins. The plasma membrane has a fluid consistency and a mosaic pattern of embedded proteins. The cell-surface membrane controls the movement of substances into and out of the cell. Cell membranes are made up of…. Phospholipids Phospholipids allow…. • Lipid- soluble substances to enter and leave the cell • Prevent water-soluble substances entering and leaving the cell • Make the membrane flexible and self-sealing. Make a note of this information Proteins are also found in the bilayer…. http://www.youtube.com/watch?v=moPJkCbKjBs&feat ure=rhttp://www.youtube.com/watch?v=moPJkCbKj Bs&feature=relatedelated http://www.youtube.com/watch?v=moPJkCbKjBs&f eature=related The fluid-mosaic model of the cell-surface membrane http://highered.mcgraw- hill.com/sites/0072495855/student_view0/chapter 2/animation__how_facilitated_diffusion_works.ht ml Movement across membranes Molecules can pass across membranes by: • Simple diffusion • Facilitated diffusion • Active transport The Permeability of the Plasma Membrane The plasma membrane is partially permeable. Macromolecules cannot pass through because of their size, and tiny charged molecules do not pass through the nonpolar interior of the membrane. Small, uncharged molecules such as oxygen and carbon dioxide pass through the membrane, down their concentration gradient. How molecules cross the plasma membrane Diffusion Diffusion is the passive movement of molecules from a higher to a lower concentration until equilibrium is reached. Gases move through plasma membranes by diffusion. Process of diffusion Gas exchange in the lungs occurs by diffusion Facilitated diffusion During facilitated diffusion, substances pass through a carrier protein following their concentration gradients.