Complete Chloroplast Genome of Japanese Larch (Larix Kaempferi): Insights Into Intraspecific Variation with an Isolated Northern Limit Population
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Invasive Alien Species Fact Sheet Phytophthora Ramorum
NOBANIS –Invasive Alien Species Fact Sheet Phytophthora ramorum Author of this species fact sheet: Anna Poimala and Arja Lilja, Finnish Forest Research Institute, Vantaa Research Unit, PO Box 18, 01301 Vantaa, Finland; +358 40 801 5377 ; [email protected] Bibliographical reference – how to cite this fact sheet: Poimala, A. & Lilja, A. (2013): NOBANIS – Invasive Alien Species Fact Sheet – Phytophthora ramorum . – From: Online Database of the European Network on Invasive Alien Species – NOBANIS www.nobanis.org , Date of access x/x/201x. Species description Scientific names: Phytophthora ramorum Werres, De Cock & Man in`t Veld, Oomycetes, Chromalveolata. Synonyms: None. Common names: Twig and leaf blight (EU), Ramorum leaf blight (North America), Sudden Oak Death= SOD (North America), tamme-äkksurm (EE), maladie de l’encre des chênes rouges (FR), mort subite du chêne (FR), tammen äkkikuolema (FI), europæisk visneskimmel (DK, European isolates) / californisk visneskimmel (DK, North American isolates), Plötslig ekdöd (SE), Plötzliches eichensterben (DE), Nagła śmier ć d ębu (POL). Fig 1 . Sporangia of Phytophthora ramorum in soil extract water, photo by Arja Lilja. 1 Fig 2 . Branched dendroid-like hyphae of Phytophthora ramorum on the bottom of an agar plate, photo by Arja Lilja. Fig 3. Clamydospore of Phytophthora ramorum , photo by Arja Lilja. Species identification Phytophthora ramorum is a heterothallic species characterized by abundant production of chlamydospores and elongate, ellipsoid, deciduous sporangia. The mean sporangium length was 43.6 µm ± 5.3 with a range from 20-79 µm, and the mean sporangium width 23.9 µm ± 2.6 with a range from 12-40 µm in measurements done by Werres and Kaminski (2005). -

Analysis of Taper Functions for Larix Olgensis Using Mixed Models and TLS
Article Analysis of Taper Functions for Larix olgensis Using Mixed Models and TLS Dandan Li , Haotian Guo, Weiwei Jia * and Fan Wang Department of Forest Management, School of Forestry, Northeast Forestry University, Harbin 150040, China; [email protected] (D.L.); [email protected] (H.G.); [email protected] (F.W.) * Correspondence: [email protected]; Tel.: +86-451-8219-1215 Abstract: Terrestrial laser scanning (TLS) plays a significant role in forest resource investigation, forest parameter inversion and tree 3D model reconstruction. TLS can accurately, quickly and nondestructively obtain 3D structural information of standing trees. TLS data, rather than felled wood data, were used to construct a mixed model of the taper function based on the tree effect, and the TLS data extraction and model prediction effects were evaluated to derive the stem diameter and volume. TLS was applied to a total of 580 trees in the nine larch (Larix olgensis) forest plots, and another 30 were applied to a stem analysis in Mengjiagang. First, the diameter accuracies at different heights of the stem analysis were analyzed from the TLS data. Then, the stem analysis data and TLS data were used to establish the stem taper function and select the optimal basic model to determine a mixed model based on the tree effect. Six basic models were fitted, and the taper equation was comprehensively evaluated by various statistical metrics. Finally, the optimal mixed model of the plot was used to derive stem diameters and trunk volumes. The stem diameter accuracy obtained by TLS was >98%. The taper function fitting results of these data were approximately the same, and the optimal basic model was Kozak (2002)-II. -

Ecology and Management of Larix Forests: a Look Ahead Proceedings of an International Symposium
Ecology and Management of Larix Forests: A Look Ahead Proceedings of an International Symposium Whitefish, Montana, U.S.A. October 5-9, 1992 Compilers: Wyman C. Schmidt Kathy J. McDonald Duchesne, L. C.; Lelu, M. A; von Aderkas, P.; Charest, Klimaszewska, K 1989. Plantlet development from imma P. J. 1992. Microprojectile-mediated DNA delivery in ture zygotic embryos of hybrid larch through somatic haploid and diploid embryogenic cells of Larix spp. embryogenesis. Plant Science. 63: 95-103. Canadian Journal of Forest Research. [In press]. Klimaszewska, K; Ward, C.; Cheliak, W. M. 1992. Cryo Ellis, D. D.; McCabe, D.; McInnis, S.; Martinell, B.; preservation and plant regeneration from embryogenic Roberts, D.; McCown, B. 1991. Transformation of white cultures oflarch (Larix x eurolepis) and black spruce spruce by electrical discharge particle acceleration. In: (Picea mariana). Journal of Expermental Botany. 43: Haissing, B. E.; Kirk, T. K; Olsen, W. L.; Raffa, K F.; 73-79. Slavicek, J. M., eds. Applications of biotechnology-to Lelu, M. A; Klimaszewska, K K; Jones, C.; Ward, C.; tree culture, protection and utilization. United States von Aderkas, P.; Charest, P. J. 1992. A laboratory guide Department of Agriculture, Forest Service, Columbus, to somatic embryogenesis in spruce and larch. Petawawa OH:I02. National Forestry Institute. Information Report. Huang, Y.; Diner, AM.; Karnosky, D. F. 1991. Agrobacter PI-X-Ul (submitted for publication). ium rhizogenes-mediated genetic transformation and von Aderkas, P.; Klimaszewska, K K; Bonga, J . M. 1990. regeneration of a conifer: oorix decidua. In: Vitro Cell. Diploid and haploid embryogenesis in Larix leptolepis, Dev. BioI. 27P: 201-207. -

Botsad 03 2018.Indd
БЮЛЛЕТЕНЬ ГЛАВНОГО БОТАНИЧЕСКОГО САДА 3/2018 (Выпуск 204) ISSN: 0366-502Х СОДЕРЖАНИЕ Учредители: Федеральное государственное ИНТРОДУКЦИЯ И АККЛИМАТИЗАЦИЯ бюджетное учреждение науки Главный ботанический сад им. Н.В. Цицина РАН ООО «Научтехлитиздат»; ООО «Мир журналов». Издатель: Фирсов Г.А. ООО «Научтехлитиздат» Журнал зарегистрирован федеральной службой по надзору в сфере связи Представители рода тисс (Taxus L.) в Ботаническом саду Петра Великого .........3 информационных технологий и массовых коммуникаций (Роскомнадзор). Шейко В.В. Свидетельство о регистрации СМИ ПИ № ФС77-46435 Lonicera chamissoi Bunge ex P.Kir. в природе и культуре ......................................12 Подписные индексы ОАО «Роспечать» 83164 «Пресса России» 11184 Волчанская А.В., Фирсов Г.А. Главный редактор: Демидов А.С., доктор биологических Долговечность и устойчивость редких древесных растений флоры наук, профессор, Россия Редакционная коллегия: России в Ботаническом саду Петра Великого .......................................................19 Бондорина И.А. доктор биол. наук, Россия Виноградова Ю.К. доктор биол. наук Россия Горбунов Ю.Н. доктор биол. наук, Сахарова С.Г., Орлова Л.В., Тарасевич В.Ф. (зам. гл. редактора), Россия Иманбаева А.А. канд. биол. наук, Казахстан Молканова О.И. канд. с/х наук, Россия К уточнению таксономии видов коллекции ботанического сада Плотникова Л.С. доктор биол. наук, проф. Россия Решетников В.Н. доктор биол. наук, СПбГЛТА (на примере Pseudolarix amabilis (J. Nelson) Rehder )..........................27 проф., Беларусь Романов М.С. канд.биол.наук, Россия Семихов В.Ф. доктор биол.наук, проф. Россия Кабанов А.В. Ткаченко О.Б. доктор биол. наук, Россия Шатко В.Г. канд. биол. наук (отв. секретарь), Россия Особенности формирования коллекции астильбы в ГБС РАН ............................40 Швецов А.Н. канд. биол. наук, Россия Huang Hongwen Prof., China Peter Wyse Jackson Dr., Prof.,USA Бугаев В.В. -
IUCN Red List of Threatened Species™ to Identify the Level of Threat to Plants
Ex-Situ Conservation at Scott Arboretum Public gardens and arboreta are more than just pretty places. They serve as an insurance policy for the future through their well managed ex situ collections. Ex situ conservation focuses on safeguarding species by keeping them in places such as seed banks or living collections. In situ means "on site", so in situ conservation is the conservation of species diversity within normal and natural habitats and ecosystems. The Scott Arboretum is a member of Botanical Gardens Conservation International (BGCI), which works with botanic gardens around the world and other conservation partners to secure plant diversity for the benefit of people and the planet. The aim of BGCI is to ensure that threatened species are secure in botanic garden collections as an insurance policy against loss in the wild. Their work encompasses supporting botanic garden development where this is needed and addressing capacity building needs. They support ex situ conservation for priority species, with a focus on linking ex situ conservation with species conservation in natural habitats and they work with botanic gardens on the development and implementation of habitat restoration and education projects. BGCI uses the IUCN Red List of Threatened Species™ to identify the level of threat to plants. In-depth analyses of the data contained in the IUCN, the International Union for Conservation of Nature, Red List are published periodically (usually at least once every four years). The results from the analysis of the data contained in the 2008 update of the IUCN Red List are published in The 2008 Review of the IUCN Red List of Threatened Species; see www.iucn.org/redlist for further details. -

Determination of the Most Effective Design for the Measurement Of
www.nature.com/scientificreports OPEN Determination of the most efective design for the measurement of photosynthetic light‑response curves for planted Larix olgensis trees Qiang Liu1,2, Weiwei Jia1* & Fengri Li 1* A photosynthetic light‑response (PLR) curve is a mathematical description of a single biochemical process and has been widely applied in many eco‑physiological models. To date, many PLR measurement designs have been suggested, although their diferences have rarely been explored, and the most efective design has not been determined. In this study, we measured three types of PLR curves (High, Middle and Low) from planted Larix olgensis trees by setting 31 photosynthetically active radiation (PAR) gradients. More than 530 million designs with diferent combinations of PAR gradients from 5 to 30 measured points were conducted to ft each of the three types of PLR curves. The infuence of diferent PLR measurement designs on the goodness of ft of the PLR curves and the accuracy of the estimated photosynthetic indicators were analysed, and the optimal design was determined. The results showed that the measurement designs with fewer PAR gradients generally resulted in worse predicted accuracy for the photosynthetic indicators. However, the accuracy increased and remained stable when more than ten measurement points were used for the PAR gradients. The mean percent error (M%E) of the estimated maximum net photosynthetic rate (Pmax) and dark respiratory rate (Rd) for the designs with less than ten measurement points were, on average, 16.4 times and 20.1 times greater than those for the designs with more than ten measurement points. -

Fluorescent Band Pattern of Chromosomes in Pseudolarix Amabilis, Pinaceae
© 2015 The Japan Mendel Society Cytologia 80(2): 151–157 Fluorescent Band Pattern of Chromosomes in Pseudolarix amabilis, Pinaceae Masahiro Hizume* Faculty of Education, Ehime University, 3 Bunkyo-cho, Matsuyama, Ehime 790–8577, Japan Received October 27, 2014; accepted November 18, 2014 Summary Pseudolarix amabilis belongs to one of three monotypic genera in Pinaceae. This species had 2n=44 chromosomes in somatic cells and its karyotype was composed of four long submetacentric chromosomes and 40 short telocentric chromosomes that varied gradually in length, supporting previous reports by conventional staining. The chromosomes were stained sequentially with the fluorochromes, chromomycin A3 (CMA) and 4′,6-diamidino-2-phenylindole (DAPI). CMA- bands appeared on 12 chromosomes at near terminal region and proximal region. DAPI-bands appeared at centromeric terminal regions of all 40 telocentric chromosomes. The fluorescent-banded karyotype of this species was compared with those of other Pinaceae genera considering taxonomical treatment and molecular phylogenetic analyses reported. On the basis of the fluorescent-banded karyotype, the relationship between Pseudolarix amabilis and other Pinaceae genera was discussed. Key words Chromomycin, Chromosome, DAPI, Fluorescent banding, Pinaceae, Pseudolarix amabilis. In Pinaceae, 11 genera with about 220 species are distinguished and grow mostly in the Northern Hemisphere (Farjon 1990). Most genera are evergreen trees, and only Larix and Pseudolarix are deciduous. Pinus is the largest genus in species number, and Cathaya, Nothotsuga and Pseudolarix are monotypic genera. The taxonomy of Pinaceae with 11 genera is complicated, having some problems in species or variety level. Several higher taxonomic treatments were reported on the base of anatomy and morphology such as resin canal in the vascular cylinder, seed scale, position of mature cones, male strobili in clusters from a single bud, and molecular characters in base sequences of several DNA regions. -
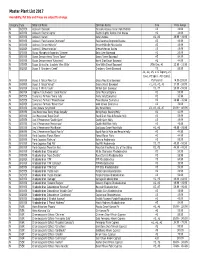
Master Plant List 2017.Xlsx
Master Plant List 2017 Availability, Pot Size and Prices are subject to change. Category Type Botanical Name Common Name Size Price Range N BREVER Azalea X 'Cascade' Cascade Azalea (Glenn Dale Hybrid) #3 49.99 N BREVER Azalea X 'Electric Lights' Electric Lights Double Pink Azalea #2 44.99 N BREVER Azalea X 'Karen' Karen Azalea #2, #3 39.99 - 49.99 N BREVER Azalea X 'Poukhanense Improved' Poukhanense Improved Azalea #3 49.99 N BREVER Azalea X 'Renee Michelle' Renee Michelle Pink Azalea #3 49.99 N BREVER Azalea X 'Stewartstonian' Stewartstonian Azalea #3 49.99 N BREVER Buxus Microphylla Japonica "Gregem' Baby Gem Boxwood #2 29.99 N BREVER Buxus Sempervirens 'Green Tower' Green Tower Boxwood #5 64.99 N BREVER Buxus Sempervirens 'Katerberg' North Star Dwarf Boxwood #2 44.99 N BREVER Buxus Sinica Var. Insularis 'Wee Willie' Wee Willie Dwarf Boxwood Little One, #1 13.99 - 21.99 N BREVER Buxus X 'Cranberry Creek' Cranberry Creek Boxwood #3 89.99 #1, #2, #5, #15 Topiary, #5 Cone, #5 Spiral, #10 Spiral, N BREVER Buxus X 'Green Mountain' Green Mountain Boxwood #5 Pyramid 14.99-299.99 N BREVER Buxus X 'Green Velvet' Green Velvet Boxwood #1, #2, #3, #5 17.99 - 59.99 N BREVER Buxus X 'Winter Gem' Winter Gem Boxwood #5, #7 59.99 - 99.99 N BREVER Daphne X Burkwoodii 'Carol Mackie' Carol Mackie Daphne #2 59.99 N BREVER Euonymus Fortunei 'Ivory Jade' Ivory Jade Euonymus #2 35.99 N BREVER Euonymus Fortunei 'Moonshadow' Moonshadow Euonymus #2 29.99 - 35.99 N BREVER Euonymus Fortunei 'Rosemrtwo' Gold Splash Euonymus #2 39.99 N BREVER Ilex Crenata 'Sky Pencil' -

Variations and Determinants of Carbon Content in Plants: a Global Synthesis
1 SUPPORTING INFORMATION 2 Variations and determinants of carbon content in plants: 3 a global synthesis 4 Suhui Ma1, Feng He2, Di Tian1, Dongting Zou1, Zhengbing Yan1, Yulong Yang3, 5 Tiancheng Zhou3, Kaiyue Huang3, Haihua Shen4, Jingyun Fang1* 6 1 Department of Ecology, College of Urban and Environmental Sciences, Peking University, Beijing 7 100871, China; 8 2 College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China; 9 3 College of Earth and Environmental Sciences, Lanzhou University, Lanzhou 730000, China; 10 4 State Key Laboratory of Vegetation and Environmental Change, Institute of Botany, Chinese 11 Academy of Sciences, Beijing 100093, China 12 *Corresponding author: Dr. Jingyun Fang 13 Email: [email protected] 14 This file includes: 15 Figure S1 16 Table S1 and S2 17 18 1 19 Figure S1. Changes in the species composition along the gradients of latitude, mean annual 20 temperature (MAT) and mean annual precipitation (MAP). The percentage of woody plants 21 decreased with the increasing latitude and with the decreasing MAT and MAP. Herb showed the 22 opposite trends with woody plants. Other life forms showed no significant change along latitudinal 23 and climatic gradients. 24 25 2 26 Table S1. Data sets in TRY that contributed to our global dataset of plant carbon content. 27 References cited in this table are attached below. DatasetID Contributor Dataset Name Reference 10 Joseph Craine Roots Of the World (ROW) Database Craine et al., 2005 34 Jon Lloyd The RAINFOR Plant Trait Database Fyllas -

Phytophthora Ramorum
Forest Health Pest Information Note 3 of 2021 Phytophthora ramorum Updated: 01/07/2021 Background Phytophthora ramorum is a harmful pathogen which is known to have over 180 hosts, many of which are tree spe- cies. Phytophthora ramorum was first noticed in the early 1990s in plant nurseries in Europe and in forests in Cali- fornia. In mainland Europe, it causes a serious blight of ornamental plants, especially Rhododendron, Camellia and Viburnum. In North America it causes a forest disease called Sudden Oak Death, killing millions of Quercus trees in deciduous forests of California and Oregon. It also costs millions annually to the ornamental nursery industry in Europe and North America. In Ireland, Phytophthora ramorum was first detected in 2002 on imported Rhododendron and Viburnum, and in the wild in 2003 on Rhododendron ponticum. In 2010, Phytophthora ramorum was found to be infecting trees in Ire- land, in particular Japanese larch (Larix kaempferi), and has since been recognised as a serious threat as it can cause damage and death of Japanese larch. Sanitation felling of infected Japanese larch stands has been carried- out – based on policy and legislative requirements, in an effort to limit the spread of the disease. By the end of 2020 Phytophthora ramorum had been found in Japanese larch at 56 forest locations. Phytophthora ramorum has also been a major problem for the United Kingdom over the last decade. There have also been limited outbreaks in north western France in Japanese larch in recent years. However, in the rest of the EU, Phytophthora ramorum has not yet proved to be a forest health issue of concern being far more associated with the horticultural nursery trade. -

Metabolic Response of Larix Olgensis A. Henry to Polyethylene Glycol-Simulated Drought Stress
Metabolic Response of Larix Olgensis A. Henry to Polyethylene Glycol-simulated Drought Stress Lei Zhang State Key Laboratory of Tree Genetics and Breeding(Northeast Forestry University) ShanShan Yan State Key Laboratory of Tree Genetics and Breeding(Northeast Forestry University) Sufang Zhang State Key Laboratory of Tree Genetics and BreedingNortheast Forestry University Pingyu Yan State Key Laboratory of Tree Genetics and Breeding(Northeast Forestry University) Junhui Wang State Key Laboratory of Tree Genetics and Breeding(Chinese Academy Of Forestry) Hanguo Zhang ( [email protected] ) State Key Laboratory of tree Genetic and breeding https://orcid.org/0000-0002-8904-539X Research article Keywords: Drought stress, Larix olgensis, differentially expressed, metabolism Posted Date: November 24th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-107824/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/28 Abstract Background Drought stress in trees limits their growth, survival, and productivity, and it negatively affects the afforestation survival rate. Molecular responses to drought stress have been extensively studied in broad- leaved species, but studies on coniferous species are limited. Results Our study focused on the molecular responses to drought stress in a coniferous species, Larix olgensis A. Henry. Drought stress was simulated in one-year-old seedlings using 25% polyethylene glycol 6000. The drought stress response in these seedlings was assessed by analyzing select biochemical parameters, along with gene expression and metabolite proles. The soluble protein content, peroxidase activity, and malondialdehyde content of L. olgensis were signicantly changed during drought stress. Quantitative gene expression analysis identied a total of 8172 differentially expressed genes in seedlings processed after 24 h, 48 h, and 96 h of drought stress treatment. -

Hylobius Abietis
On the cover: Stand of eastern white pine (Pinus strobus) in Ottawa National Forest, Michigan. The image was modified from a photograph taken by Joseph O’Brien, USDA Forest Service. Inset: Cone from red pine (Pinus resinosa). The image was modified from a photograph taken by Paul Wray, Iowa State University. Both photographs were provided by Forestry Images (www.forestryimages.org). Edited by: R.C. Venette Northern Research Station, USDA Forest Service, St. Paul, MN The authors gratefully acknowledge partial funding provided by USDA Animal and Plant Health Inspection Service, Plant Protection and Quarantine, Center for Plant Health Science and Technology. Contributing authors E.M. Albrecht, E.E. Davis, and A.J. Walter are with the Department of Entomology, University of Minnesota, St. Paul, MN. Table of Contents Introduction......................................................................................................2 ARTHROPODS: BEETLES..................................................................................4 Chlorophorus strobilicola ...............................................................................5 Dendroctonus micans ...................................................................................11 Hylobius abietis .............................................................................................22 Hylurgops palliatus........................................................................................36 Hylurgus ligniperda .......................................................................................46