Paracetamol and Cyclooxygenase Inhibition: Is There a Cause for Concern? Burkhard Hinz,1 Kay Brune2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Issue 3 Combinations Were Banned in the Month of March, 2016
344 FDC’s REGIMEN Banned in CHIPS India The Health Ministry has banned 344 ‘Fixed HIP C S ✪ Dose Combination’ (FDC) ✪ drugs, leading to an immediate suspension of the manufacturing and sale of some popular medicines in India. Fixed drug combinations have mushroomed in the market as the Pharmaceutical companies ✪ in their quest for newer products and to increase G ✪ UNTUR FR Drug Information News Letter its sales, mix and match ingredients into a single OM C EATION molecule to market them as newer remedies. 344 of such ONCEPT TO CR Jan-Mar 2016, Volume 1, Issue 3 combinations were banned in the month of March, 2016. Keeping in view of the Risk that is associated with these Fixed drug combinations and safer alternatives being available in the market, the Ministry moved ahead with its decision to ban all the 344 fixed drug combinations. Simethicone, *fixed dose combination of Magaldrate + Papain + Fungul Here is the list of 344 banned fixed Drug Combinations in India Diastase + Simethicone, *fixed dose combination of Rabeprazole + Zinc + Domperidone, *fixed dose combination of Famotidine + Oxytacaine + *Aceclofenac + Paracetamol + Rabeprazole, *Nimesulide + Diclofenac, *Diphenoxylate + Atropine + Furazolidone, * Combikit of Fluconazole Magaldrate,*fixed dose combination of Ranitidine + Domperidone + *Nimesulide + Cetirizine + Caffeine, *Nimesulide + Tizanidine, Tablet, Azithromycin Tablet and Ornidazole Tablets, *Ciprofloxacin + Simethicone,*fixed dose combination of Alginic Acid + Sodium *Paracetamol + Cetirizine + Caffeine*Diclofenac + -

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

Ascorbic Acid/ Paracetamol/ Phenylephrine Hydrochloride PSUSA/00000255/202006 List of Nationally Authorised Medicinal Products
11 February 2011 Human Medicines Evaluation Division List of nationally authorised medicinal products Active substance: ascorbic acid/paracetamol/phenylephrine hydrochloride Procedure no.: PSUSA/00000255/202006 Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2021. Reproduction is authorised provided the source is acknowledged. Member State where Product full name MRP/DCP or CP National Authorisation MAH of product in the member product is (in authorisation country) Authorisation number Number state authorised OMEGA PHARMA HUNGARY Coldrex méz és citrom ízű por belsőleges oldathoz not available OGYI-T-1715/06 KFT. HU OMEGA PHARMA HUNGARY Coldrex méz és citrom ízű por belsőleges oldathoz not available OGYI-T-1715/05 KFT. HU OMEGA PHARMA HUNGARY Coldrex méz és citrom ízű por belsőleges oldathoz not available OGYI-T-1715/11 KFT. HU Blackcurrant Coldrex Powders not available PL 02855/0271 OMEGA PHARMA LTD UK AZIENDE CHIMICHE RIUNITE TACHIFLUDEC polvere per soluzione orale gusto ANGELINI FRANCESCO - menta not available 034358073 A.C.R.A.F. S.P.A. IT AZIENDE CHIMICHE RIUNITE TACHIFLUDEC polvere per soluzione orale gusto ANGELINI FRANCESCO - menta. not available 034358085 A.C.R.A.F. S.P.A. IT List of nationally authorised medicinal products Page 2/8 COLDREX MAXGRIP MENTHOL & BERRIES, 1000 mg/70 mg/10 mg, suukaudse lahuse pulber kotikeses not available 798812 RICHARD BITTNER AG, EE GLAXOSMITHKLINE CONSUMER HEALTHCARE Beechams Cold & Flu Hot Lemon not available MA 932/00103 (UK) TRADING LIMITED MT OMEGA PHARMA HUNGARY Coldrex citrom ízű por belsőleges oldathoz not available OGYI-T-1715/01 KFT. -

Management of Poisoning
MOH CLINICAL PRACTICE GUIDELINES December/2011 Management of Poisoning Health Ministry of Sciences Chapter of Emergency College of College of Family Manpower Authority Physicians Physicians, Physicians Academy of Medicine, Singapore Singapore Singapore Singapore Medical Pharmaceutical Society Society for Emergency Toxicology Singapore Paediatric Association of Singapore Medicine in Singapore Society (Singapore) Society Executive summary of recommendations Details of recommendations can be found in the main text at the pages indicated. Principles of management of acute poisoning – resuscitating the poisoned patient GPP In a critically poisoned patient, measures beyond standard resuscitative protocol like those listed above need to be implemented and a specialist experienced in poisoning management should be consulted (pg 55). GPP D Prolonged resuscitation should be attempted in drug-induced cardiac arrest (pg 55). Grade D, Level 3 1 C Titrated doses of naloxone, together with bag-valve-mask ventilation, should be administered for suspected opioid-induced coma, prior to intubation for respiratory insuffi ciency (pg 56). Grade C, Level 2+ D In bradycardia due to calcium channel or beta-blocker toxicity that is refractory to conventional vasopressor therapy, intravenous calcium, glucagon or insulin should be used (pg 57). Grade D, Level 3 B Patients with actual or potential life threatening cardiac arrhythmia, hyperkalaemia or rapidly progressive toxicity from digoxin poisoning should be treated with digoxin-specifi c antibodies (pg 57). Grade B, Level 2++ B Titrated doses of benzodiazepine should be given in hyperadrenergic- induced tachycardia states resulting from poisoning (pg 57). Grade B, Level 1+ D Non-selective beta-blockers, like propranolol, should be avoided in stimulant toxicity as unopposed alpha agonism may worsen accompanying hypertension (pg 57). -

PRODUCT INFORMATION Mefenamic Acid Item No
PRODUCT INFORMATION Mefenamic Acid Item No. 23650 CAS Registry No.: 61-68-7 OH Formal Name: 2-[(2,3-dimethylphenyl)amino]-benzoic acid O Synonyms: C.I. 473, CN 35355, NSC 94437 H C H NO MF: 15 15 2 N FW: 241.3 Purity: ≥98% Supplied as: A solid Storage: 4°C Stability: ≥1 year Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Mefenamic acid is supplied as a solid. A stock solution may be made by dissolving the mefenamic acid in the solvent of choice, which should be purged with an inert gas. Mefenamic acid is slightly soluble in DMSO and methanol. Description Mefenamic acid is a non-steroidal anti-inflammatory drug (NSAID) and a COX-2 inhibitor.1 It binds to COX-2 (Kd = 4 nM) and inhibits COX-2-dependent oxygenation of arachidonic acid (Item Nos. 90010 | 90010.1 | 10006607) in vitro (Ki = 10 µM). Mefenamic acid (30 mg/kg) reduces acetic acid-induced writhing in rats.2 It inhibits increases in skin thickness in a mouse model of delayed-type hypersensitivity induced by dinitrochlorobenzene (DNBC).2 Mefenamic acid also reduces the antibody response to sheep red blood cells in mice. References 1. Prusakiewicz, J.J., Duggan, K.C., Rouzer, C.A., et al. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 48(31), 7353-7355 (2009). 2. Roszkowski, A.P., Rooks, W.H., Tomolonis, A.J., et al. Anti-inflammatory and analgetic properties of d-2-(6’-methoxy-2’-naphthyl)-propionic acid (naproxen). -

Open Thesis Master Document V5.0.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Science IDENTIFICATION OF ENDOGENOUS MODULATORS FOR THE ARYL HYDROCARBON RECEPTOR A Thesis in Genetics by Christopher R. Chiaro © 2007 Christopher R. Chiaro Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December, 2007 The thesis of Christopher R. Chiaro was reviewed and approved* by the following: Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Thesis Advisor Chair of Committee C. Channa Reddy Distinguished Professor of Veterinary Science A. Daniel Jones Senior Scientist Department of Chemistry John P. Vanden Heuvel Professor of Veterinary Science Richard Ordway Associate Professor of Biology Chair of Genetics Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor capable of being regulated by a structurally diverse array of chemicals ranging from environmental carcinogens to dietary metabolites. A member of the basic helix-loop- helix/ Per-Arnt-Sim (bHLH-PAS) super-family of DNA binding regulatory proteins, the AhR is an important developmental regulator that can be detected in nearly all mammalian tissues. Prior to ligand activation, the AhR resides in the cytosol as part of an inactive oligomeric protein complex comprised of the AhR ligand-binding subunit, a dimer of the 90 kDa heat shock protein, and a single molecule each of the immunophilin like X-associated protein 2 (XAP2) and p23 proteins. Functioning as chemosensor, the AhR responds to both endobiotic and xenobiotic derived chemical ligands by ultimately directing the expression of metabolically important target genes. -

N-Acyl-Dopamines: Novel Synthetic CB1 Cannabinoid-Receptor Ligands
Biochem. J. (2000) 351, 817–824 (Printed in Great Britain) 817 N-acyl-dopamines: novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo Tiziana BISOGNO*, Dominique MELCK*, Mikhail Yu. BOBROV†, Natalia M. GRETSKAYA†, Vladimir V. BEZUGLOV†, Luciano DE PETROCELLIS‡ and Vincenzo DI MARZO*1 *Istituto per la Chimica di Molecole di Interesse Biologico, C.N.R., Via Toiano 6, 80072 Arco Felice, Napoli, Italy, †Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, R. A. S., 16/10 Miklukho-Maklaya Str., 117871 Moscow GSP7, Russia, and ‡Istituto di Cibernetica, C.N.R., Via Toiano 6, 80072 Arco Felice, Napoli, Italy We reported previously that synthetic amides of polyunsaturated selectivity for the anandamide transporter over FAAH. AA-DA fatty acids with bioactive amines can result in substances that (0.1–10 µM) did not displace D1 and D2 dopamine-receptor interact with proteins of the endogenous cannabinoid system high-affinity ligands from rat brain membranes, thus suggesting (ECS). Here we synthesized a series of N-acyl-dopamines that this compound has little affinity for these receptors. AA-DA (NADAs) and studied their effects on the anandamide membrane was more potent and efficacious than anandamide as a CB" transporter, the anandamide amidohydrolase (fatty acid amide agonist, as assessed by measuring the stimulatory effect on intra- hydrolase, FAAH) and the two cannabinoid receptor subtypes, cellular Ca#+ mobilization in undifferentiated N18TG2 neuro- CB" and CB#. NADAs competitively inhibited FAAH from blastoma cells. This effect of AA-DA was counteracted by the l µ N18TG2 cells (IC&! 19–100 M), as well as the binding of the CB" antagonist SR141716A. -

1. Generic Name Paracetamol, Phenylephrine, Chlorpheniramine Maleate, Sodium Citrate, Menthol 2. Qualitative and Quantitative Co
1. Generic Name Paracetamol, Phenylephrine, Chlorpheniramine maleate, Sodium citrate, Menthol 2. Qualitative and Quantitative composition Paracetamol 125 mg Phenylephrine 5 mg Chlorpheniramine maleate 1 mg Sodium citrate 60 mg Mentholated flavoured syrupy base q.s. 3. Dosage form and strength Oral syrup containing Paracetamol 125 mg, Phenylephrine 5 mg, Chlorpheniramine maleate 1 mg. 4. Clinical particulars 4.1 Therapeutic indication Sinarest Syrup is indicated in children below 40 kg weight for: • Relief of nasal and sinus congestion. • Relief of allergic symptoms of the nose or throat due to upper respiratory tract allergies. • Relief of sinus pain and headache. • Adjunct with antibacterial in sinusitis, tonsillitis and otitis media. 4.2 Posology and method of administration Children of age between 2 to 12 years (body weight- 6 - 40 kg): The usual recommended dose is as shown in the table below which will be given to the patient: Weight Age Dose 6 – 22.9 2 – 7 years 5 ml BID 11.9 – 40 7 – 12 years 5 ml TID 4.3 Contraindication The use of Sinarest syrup is contraindicated in patients with: Hypersensitivity to any of the ingredients of the formulation. Severe hypertension. 4.4 Special warnings and precautions for use In case a hypersensitivity reaction occurs which is rare, Sinarest syrup should be discontinued. Sinarest syrup contains Paracetamol and therefore should not be used in conjunction with other Paracetamol containing products. Sinarest syrup should be used with caution in patients with renal or hepatic dysfunction, diabetes mellitus, hyperthyroidism, cardiovascular problems, epilepsy and closed angle glaucoma. 4.5 Drug interactions Clinically significant drug interactions may occur on concomitant administration of Sinarest syrup with monoamine oxidase inhibitors, tricyclic antidepressants, beta-adrenergic agents, and methyldopa, reserpine and veratrum alkaloids. -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

THE L5178Y SUBLINES: a SUMMARY of 40 YEAR STUDIES in WARSAW Irena Szumiel
ISSN 1425-7351 PL0601824 RAPORTY IChTJ. SERIA B nr 2/2005 THE L5178Y SUBLINES: A SUMMARY OF 40 YEAR STUDIES IN WARSAW Irena Szumiel INSTYTUT CHEMII I TECHNIKI JĄDROWEJ INSTITUTE OF NUCLEAR CHEMISTRY AND TECHNOLOGY RAPORTY IChTJ. SERIA B nr 2/2005 THE L5178Y SUBLINES: A SUMMARY OF 40 YEAR STUDIES IN WARSAW Irena Szumiel Warszawa 2005 AUTHOR Irena Szumiel Department of Radiobiology and Health Protection, Institute of Nuclear Chemistry and Technology This report is an extended version of the paper: " 'The L5178Y sublines: a look back from 40 years. 1.General characteristics. 2. Response to ionising radiation", published in International Journal of Radiation Biology, vol. 81 (2005) by kind permission of Taylor & Francis (http://www. tandf.co.uk) ADDRESS OF THE EDITORIAL OFFICE Institute of Nuclear Chemistry and Technology Dorodna 16, 03-195 Warszawa, POLAND phone: (+4822) 811 06 56, fax: (+4822) 81115 32, e-mail: [email protected] Papers are published in the form as received from the Authors The L5178Y sublines: a summary of 40 year studies in Warsaw The purpose of this report has been to review and summarise the results of 40 year studies concerning the general characteristics and response to UVC radiation, hydrogen peroxide and ionising radiation of the pair of L5178Y (LY) sublines, LY-R and LY-S, that differ in sen- sitivity to various DNA damaging agents. Comparison of karyotypes shows a number of differences in the banding patterns. Differences are found in ion transport and the ganglioside pattern of the plasma membranes, as well as in the content and turnover rate of poly(ADP-ribose) polymers. -
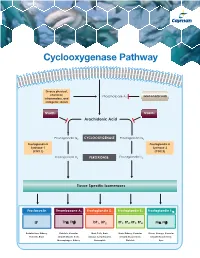
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally.