Rapid Coprecipitation Technique Using Yttrium Hydroxide for the Preconcentration and Separation of Trace Elements in Saline Water Prior to Their ICP-AES Determination
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Improvement of High−Temperature Characteristics of the Sintered
686 EJ60ケooho勿zゑsψりノ 一Article一 Improvement of High-Temperature Characteristics of the Sintered Nickel Positive Electrode for an Alkaline Storage Battery Katsuhiko SHINYAMA,*Yoshifumi MAGARI,Atsuhiro FUNAHASHI, Toshiyuki NOHMA,and Ikuo YONEZU R&D Business Unit,Mobile Energy Company,Sanyo Electric Co.,Ltd.(7-3-21bukidai-higashimachi,Nishi-ku,Kobe City,Hyogo651-2242,Japan) Received August9,2002;Accepted May20,2003 The high-temperature characteristics of sintered nickel positive electrodes for alkaline storage batteries such as nickel・metal hydride batteries and nicke1-cadmium batteries were investigated.Generally,the discharge capacity of an alkaline storage battery charged at high temperature is smaller than that charged at room temperature due to the oxygen evolution reaction.In order to enhance high-temperature characteristics,we coated sintered n量ckel positive electrodes with yttrium hydroxide,calcium hydroxide or cobalt hydroxide.Th6high-temperature characteristics of the sintered nickel positive electrodes coated with y血ium hydroxide and calcium hydroxide by immersing them in sodium hydroxide solution after immersing in nitrate solution were greatly enhanced because of the increased oxy. gen OVerVOltage. κ¢y防鳩:Sintered Nickel Positive Electrode,High-temperature Characteristics,Alkaline Storage Battery,Oxygen Overvoltage l lntroduction In the past,the electrolyte composition3)and dissolv- Cun℃ntly,alkaline storage batteries such as nicke1- ing such elements as cobalt and calcium into nickel hy- cadmium batteries and nickel-metal hydride -

Evaluation of Anti-Corrosion and Anti-Galling Performance
EVALUATION OF ANTI-CORROSION AND ANTI-GALLING PERFORMANCE OF A NOVEL GREASE COMPOUND A Thesis by JOHN OLUMIDE REIS Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Hong Liang Committee Members, Chii-Der Suh Sevan Goenezan Head of Department, Andreas A. Polycarpou December 2017 Major Subject: Mechanical Engineering Copyright 2017 John Olumide Reis ABSTRACT In this research, the effectiveness of CeO2, Y2O3 and Al2O3 as anti-corrosion and anti-wear additives in commercial grease was investigated. An experimental approach was used to carry on the research. The weight loss of steel coupons protected with a layer of thin grease, with and without the anti-corrosion additives in a corrosive environment was determined. The Friction factor of the grease compound was also evaluated. The accelerated corrosion tests were performed in a salt spray chamber for an exposure time of 2 weeks (336 hours). The corrosive medium was 5 % wt. of Brine. By varying the weight compositions of the additives (1% wt. and 3% wt.), and comparing the corrosion rates with that of base grease, the effectiveness of various grease additives was evaluated. The result showed that corrosive losses of the test samples can be effectively reduced by adding relatively small amounts of CeO2, Y2O3 and Al2O3 to the base grease. Corroded surfaces were examined using the Optical microscope to clarify the corrosion mechanism. The friction test was carried out using a galling tester. The standard test procedure as specified in API RP 7A1 was followed. -
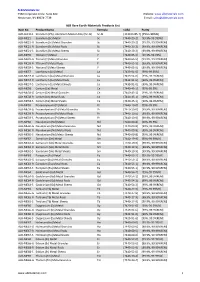
ALB Materials Inc Product List.Xlsx
ALB Materials Inc 2360 Corporate Circle. Suite 400 Website: www.albmaterials.com Henderson, NV 89074-7739 E-mail: [email protected] ALB Rare Earth Materials Products List Item No. Product Name Formula CAS# Purity ALB-AL2113 Scandium (2%)- Aluminum Master Alloy (Sc-Al) Sc-Al [113413-85-7] [2%Sc+98%Al] ALB-ME21 Scandium (Sc) Metal Sc [7440-20-2] [99.9%-99.999%] ALB-ME21-G Scandium (Sc) Metal Granules Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-R Scandium (Sc) Metal Rods Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-S Scandium (Sc) Metal Sheets Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME39 Yttrium (Y) Metal Y [7440-65-5] [99.9%-99.99%] ALB-ME39-G Yttrium (Y) Metal Granules Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-R Yttrium (Y) Metal Rods Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-S Yttrium (Y) Metal Sheets Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME57 Lanthanum (La) Metal La [7439-91-0] [99%-99.95%] ALB-ME57-G Lanthanum (La) Metal Granules La [7439-91-0] [99%, 99.9%REM] ALB-ME57-R Lanthanum (La) Metal Rods La [7439-91-0] [99%, 99.9%REM] ALB-ME57-S Lanthanum (La) Metal Sheets La [7439-91-0] [99%, 99.9%REM] ALB-ME58 Cerium (Ce) Metal Ce [7440-45-1] [99%-99.9%] ALB-ME58-G Cerium (Ce) Metal Granules Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-R Cerium (Ce) Metal Rods Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-S Cerium (Ce) Metal Sheets Ce [7440-45-1] [99%, 99.9%REM] ALB-ME59 Praseodymium (Pr) Metal Pr [7440-10-0] [99%-99.9%] ALB-ME59-G Praseodymium (Pr) Metal Granules Pr [7440-10-0] [99.9%, 99.99%REM] ALB-ME59-R Praseodymium (Pr) Metal Rods Pr [7440-10-0] -

Zirconia-Ceria-Yttria-Based Mixed Oxide and Process for Producing The
(19) TZZ_ _Z_T (11) EP 1 921 044 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C01G 25/00 (2006.01) B01J 23/10 (2006.01) 28.02.2018 Bulletin 2018/09 B01D 53/94 (2006.01) B01J 21/06 (2006.01) B01J 35/10 (2006.01) (21) Application number: 07118246.3 (22) Date of filing: 10.10.2007 (54) Zirconia-ceria-yttria-based mixed oxide and process for producing the same Auf Zirkonium-Cerium-Yttrium basierendes Mischoxid und Herstellungsverfahren dafür Oxyde mixte à base de zirconium-cérium-yttrium et son procédé de fabrication (84) Designated Contracting States: • Kodama, Hiroshi DE FR GB Osaka 5590025 (JP) (30) Priority: 12.10.2006 JP 2006305934 (74) Representative: Manley, Nicholas Michael 08.08.2007 JP 2007206394 WP Thompson 8th Floor (43) Date of publication of application: 1 Mann Island 14.05.2008 Bulletin 2008/20 Liverpool L3 1BP (GB) (73) Proprietor: Daiichi Kigenso Kagaku Kogyo Co., (56) References cited: Ltd. EP-A- 1 006 081 EP-A- 1 035 074 Osaka-shi, EP-A- 1 894 620 WO-A-2007/093593 Osaka 559-0025 (JP) JP-A- 2003 277 059 US-B1- 6 387 338 (72) Inventors: • Okamoto, Hiroshi Osaka 5590025 (JP) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been paid. -

Tompbnents of Fission and 'Natural Radioactivity Found in Water Are
DOCUMENT RESUME,, A ED '170 1110 SE027 bill. 0 AUTHOR Thatcher, L.L. ;And Others 1 TITLE Techniques of Water-Resources Investigations of the United. States' Geblogical.Survey. Book 5, Laboratory, Analysis. Chapter- A5, Methodscr Dete-rmination of - Radioactive Substances in Water and Fluvial Sediments. INSTITUTION -Geological Survey (Dept,: .of Interior) ,Reston, PUB DATE 77 NO Tr 95p. ;For related document, see ED 131 218 AVAILABLE FRQM Super.intendent of Documents, U.S. Government Printing Off /ce , Washington, D.C. -20402(Stock Number 024- 0.01- 02928- 6:, No price quoted) */ FDRS PRICE MF01/PC04 Plus Postage. DESCRIPTORS *ChemiCal Analysis; *Geolo'gy; *LaboratorTechniques; PhYsic; Pollution; Retource Materials; S.c ence Equipment; Water Pollution control IDENTIFIERS *RadioactiveVs- Substances; *Sediments; Water Quality ABSTkACT Analytical methods for determining important , tompbnents of fission and 'natural radioactivity found in water are reportedThe discussion of each method includes conditions for application of the method, a summary of the method, interferences, required apparatus, Procedures, calculations and estimation of precision. Isotopes considered are cesium-137, strontium-90, lead-210, radium-226. and 228, tritium, and carbon-714. `When two methods are in use for an isotope,- both methods are reported. Techniques for the collection and preservation of water samples to be anallze?d for radioactivity are also discuSsed.(CS) v. *********************************************************************** 4 Re. productions supplied -

Interagency Committee on Chemical Management
DECEMBER 14, 2018 INTERAGENCY COMMITTEE ON CHEMICAL MANAGEMENT EXECUTIVE ORDER NO. 13-17 REPORT TO THE GOVERNOR WALKE, PETER Table of Contents Executive Summary ...................................................................................................................... 2 I. Introduction .......................................................................................................................... 3 II. Recommended Statutory Amendments or Regulatory Changes to Existing Recordkeeping and Reporting Requirements that are Required to Facilitate Assessment of Risks to Human Health and the Environment Posed by Chemical Use in the State ............................................................................................................................ 5 III. Summary of Chemical Use in the State Based on Reported Chemical Inventories....... 8 IV. Summary of Identified Risks to Human Health and the Environment from Reported Chemical Inventories ........................................................................................................... 9 V. Summary of any change under Federal Statute or Rule affecting the Regulation of Chemicals in the State ....................................................................................................... 12 VI. Recommended Legislative or Regulatory Action to Reduce Risks to Human Health and the Environment from Regulated and Unregulated Chemicals of Emerging Concern .............................................................................................................................. -

Coprecipitation of Yttrium and Aluminium Hydroxide for Preparation of Yttrium Aluminium Garnet
Coprecipitation of yttrium and aluminium hydroxide for preparation of yttrium aluminium garnet Citation for published version (APA): Vrolijk, J. W. G. A., Willems, J. W. M. M., & Metselaar, R. (1990). Coprecipitation of yttrium and aluminium hydroxide for preparation of yttrium aluminium garnet. Journal of the European Ceramic Society, 6(1), 47-51. https://doi.org/10.1016/0955-2219(90)90034-D DOI: 10.1016/0955-2219(90)90034-D Document status and date: Published: 01/01/1990 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. -

United States Patent (19) 11 Patent Number: 6,077,625 Yano Et Al
USOO6077625A United States Patent (19) 11 Patent Number: 6,077,625 Yano et al. (45) Date of Patent: Jun. 20, 2000 54) NON-SINTERED NICKEL ELECTRODE FOR 0757395A1 2/1997 European Pat. Off.. ALKALINE STORAGE BATTERY 62-234867 10/1987 Japan. 1-272050 10/1989 Japan. 75 Inventors: Mutsumi Yano, Hirakata; Mitsunori 3-078965 4/1991 Japan. Tokuda, Osaka, Kousuke Satoguchi, 5-028992 2/1993 Japan. Tokushima; Shin Fujitani; Koji Nishio, WO both of Hirakata, all of Japan 96.14666A1 5/1996 WIPO. 73 Assignee: Sanyo Electric Co., Ltd., Osaka, Japan Primary Examiner Bruce F. Bell Attorney, Agent, or Firm-Birch, Stewart, Kolasch & Birch, 21 Appl. No.: 09/097,679 LLP 22 Filed: Jun. 16, 1998 57 ABSTRACT 30 Foreign Application Priority Data Non-sintered nickel electrodes for alkaline Storage batteries which can express high active material utilization efficiency Jun. 16, 1997 JP Japan .................................... 9-176314 not only at the time of charging at ordinary temperature but Jun. 16, 1997 JP Japan .................................... 9-176315 also at the time of charging in a high-temperature atmo Sphere are provided by using an active material powder (51) Int. Cl. ................................................ HO1 M 4/32 composed of composite particles each comprising a Sub 52 U.S. Cl. ......................... 429/223; 429/59; 429/218.2; Strate particle containing nickel hydroxide, an inner coat 204/290 R layer covering the Substrate particle and comprising yttrium, 58 Field of Search .......................... 429/223, 59, 218.2; Scandium or a lanthanoid, or an yttrium, Scandium or 204/290 R lanthanoid compound, and an outer coat layer covering the inner coat layer and comprising cobalt or a cobalt 56) References Cited compound, or composed of composite particles each com U.S. -

The Thermodynamic Properties of Neodymium Hydroxide In
THE THERMODYNAMIC PROPERTIES OF NEODYMIUM HYDROXIDE IN ACID, NEUTRAL, AND ALKALINE SOLUTIONS AT 25°C.: AN INTERPRETATION OF THE IONIC SPECIES PRESENT IN AQUEOUS SOLUTIONS OF NEODYMIUM SALTS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By RUSSELL STUART TOBIAS, B. Sc. 4 \ «V St i't The Ohio State University 1956 Approved by: Adviser Department! of Chemistry AC KNOWLEDGEMENT The author wishes to express his sincere appreciation to Professor A. B. Garrett for his encouragement and counsel during the course of this investigation. I also wish to thank him for his interest in my welfare while I have been a student at The Ohio State University. I also wish to thank The Ohio State University for the assistant ship granted to me and the Visking Corporation, the General Electric Corporation, E. I. du Pont de Nemours and Company, and the Allied Chemical and Dye Company for financial assistance during the course of my studies. Finally, I would like to acknowledge the assistance of Mr. G. W. Leddicotte, of the Oak Ridge National Labora tory, who performed the activation analyses described in this work. TABLE OF CONTENTS Subject Page INTRODUCTION ............................................. 1 HISTORICAL REVIEW ........................................ 3 The Basicity of the Rare Earth Oxides ............ 3 The System Neodymium Oxide-Water ................. 4 The Hydrolysis of Neodymium Solutions .......... 9 The Analyses of Solutions of Neodymium ........... 11 EXPERIMENTAL . .. 18 General Procedure ................................... 18 Preparation of the Reagents ....................... 18 Neodymium Oxide ................................. 18 Hydrochloric Acid Solutions ................... 20 Sodium Hydroxide Solutions .......... 20 Perchloric Acid Solutions .......... -

Toxicological Profile for Strontium
TOXICOLOGICAL PROFILE FOR STRONTIUM U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry April 2004 STRONTIUM ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. STRONTIUM iii UPDATE STATEMENT A Toxicological Profile for strontium, Draft for Public Comment was released in July 2001. This edition supersedes any previously released draft or final profile. Toxicological profiles are revised and republished as necessary. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, Mailstop F-32 Atlanta, Georgia 30333 vi Background Information The toxicological profiles are developed by ATSDR pursuant to Section 104(i) (3) and (5) of the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund) for hazardous substances found at Department of Energy (DOE) waste sites. CERCLA directs ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List (NPL) and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. ATSDR and DOE entered into a Memorandum of Understanding on November 4, 1992 which provided that ATSDR would prepare toxicological profiles for hazardous substances based upon ATSDR=s or DOE=s identification of need. The current ATSDR priority list of hazardous substances at DOE NPL sites was announced in the Federal Register on July 24, 1996 (61 FR 38451). -

Preparation and Photoluminescence of Yttrium Hydroxide and Yttriumoxide Doped with Europiumnanowires
ARTICLE IN PRESS Journal of Crystal Growth 277 (2005) 643–649 www.elsevier.com/locate/jcrysgro Preparation and photoluminescence of yttrium hydroxide and yttriumoxide doped with europiumnanowires Xingcai Wua,b,Ã, Yourong Taoa, Fei Gaoa, Lin Donga, Zheng Hua aDepartment of Chemistry, Laboratory of Mesoscopic Materials Science, Nanjing University, Nanjing 210093, People’s Republic of China bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, People’s Republic of China Received 17 July 2004; accepted 27 January 2005 Available online 10 March 2005 Communicated by J.M. Redwing Abstract Single-crystalline Y(OH)3:Eu nanowires have been prepared by polymer-assisted hydrothermal method. The yields of the nanowires were higher than 95%. XRD patterns showed that the products were the hexagonal Y(OH)3 structure. SEM images displayed that the nanowires consisted of nanobelts and nanorods. The nanorods have the diameter of about 85 nmand length up to several tens of microns,and the nanobelts have the width of 95 nm, and the thickness of 20 nm, and the length of several tens of microns. Y(OH)3:Eu nanowires were converted to Y2O3:Eu nanowires by thermal decomposition at 500 1C for 3 h. The morphologies still remained. The photoluminescence (PL) properties of Y(OH)3:Eu and Y2O3:Eu nanowires were studied. There are four strong peaks at 592, 616, 651 and 696 nmfor the Y(OH)3:Eu nanowires under the excitation of 396 nm. There is only strong peak at 611 nm for the Y2O3:Eu nanowires under the excitation of 465 nm. r 2005 Elsevier B.V. -

HYSYS OLI Interface
HYSYS® 2004.2 OLI Interface Reference Guide Copyright October 2005 Copyright © 1981-2005 by Aspen Technology, Inc. All rights reserved. Aspen Accounting.21™, Aspen ACM Model Export, Aspen ACOL™, Aspen ACX™ Upgrade to ACOL™, Aspen Adsim®, Aspen Advisor™, Aspen Aerotran®, Aspen Alarm & Event™, Aspen APLE™, Aspen Apollo™, Aspen AtOMS™, Aspen Batch and Event Extractor, Aspen Batch Plus®, Aspen Batch.21™, Aspen Batch.21™ CBT, Aspen BatchCAD™, Aspen BatchSep™, Aspen Blend Model Library™, Aspen Blend™, Aspen BP Crude Oil Database, Aspen Calc CBT, Aspen Calc™, Aspen Capable-to-Promise®, Aspen CatRef®, Aspen Chromatography®, Aspen Cim-IO Core™, Aspen Cim-IO™ for @AGlance, Aspen Cim-IO™ for ABB 1180/ 1190 via DIU, Aspen Cim-IO™ for Bailey SemAPI, Aspen Cim-IO™ for DDE, Aspen Cim-IO™ for Eurotherm Gauge via DCP, Aspen Cim-IO™ for Fisher-Rosemount Chip, Aspen Cim-IO™ for Fisher-Rosemount RNI, Aspen Cim-IO™ for Foxboro FOXAPI, Aspen Cim-IO™ for G2, Aspen Cim-IO™ for GE FANUC via HCT, Aspen Cim-IO™ for Hitachi Ex Series, Aspen Cim-IO™ for Honeywell TDC 3000 via HTL/access, Aspen Cim-IO™ for Intellution Fix, Aspen Cim-IO™ for Measurex MCN, Aspen Cim-IO™ for Measurex ODX, Aspen Cim-IO™ for Moore Apacs via Nim (RNI), Aspen Cim-IO™ for OPC, Aspen Cim-IO™ for PI, Aspen Cim- IO™ for RSLinx, Aspen Cim-IO™ for SetCim/InfoPlus-X/InfoPlus.21, Aspen Cim-IO™ for Toshiba Tosdic, Aspen Cim-IO™ for ULMA 3D, Aspen Cim-IO™ for Westinghouse, Aspen Cim-IO™ for WonderWare InTouch, Aspen Cim-IO™ for Yokogawa ACG10S, Aspen Cim-IO™ for Yokogawa EW3, Aspen Collaborative Forecasting™,