Tompbnents of Fission and 'Natural Radioactivity Found in Water Are
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Improvement of High−Temperature Characteristics of the Sintered
686 EJ60ケooho勿zゑsψりノ 一Article一 Improvement of High-Temperature Characteristics of the Sintered Nickel Positive Electrode for an Alkaline Storage Battery Katsuhiko SHINYAMA,*Yoshifumi MAGARI,Atsuhiro FUNAHASHI, Toshiyuki NOHMA,and Ikuo YONEZU R&D Business Unit,Mobile Energy Company,Sanyo Electric Co.,Ltd.(7-3-21bukidai-higashimachi,Nishi-ku,Kobe City,Hyogo651-2242,Japan) Received August9,2002;Accepted May20,2003 The high-temperature characteristics of sintered nickel positive electrodes for alkaline storage batteries such as nickel・metal hydride batteries and nicke1-cadmium batteries were investigated.Generally,the discharge capacity of an alkaline storage battery charged at high temperature is smaller than that charged at room temperature due to the oxygen evolution reaction.In order to enhance high-temperature characteristics,we coated sintered n量ckel positive electrodes with yttrium hydroxide,calcium hydroxide or cobalt hydroxide.Th6high-temperature characteristics of the sintered nickel positive electrodes coated with y血ium hydroxide and calcium hydroxide by immersing them in sodium hydroxide solution after immersing in nitrate solution were greatly enhanced because of the increased oxy. gen OVerVOltage. κ¢y防鳩:Sintered Nickel Positive Electrode,High-temperature Characteristics,Alkaline Storage Battery,Oxygen Overvoltage l lntroduction In the past,the electrolyte composition3)and dissolv- Cun℃ntly,alkaline storage batteries such as nicke1- ing such elements as cobalt and calcium into nickel hy- cadmium batteries and nickel-metal hydride -

Rapid Coprecipitation Technique Using Yttrium Hydroxide for the Preconcentration and Separation of Trace Elements in Saline Water Prior to Their ICP-AES Determination
ANALYTICAL SCIENCES AUGUST 2007, VOL. 23 1021 2007 © The Japan Society for Analytical Chemistry Notes Rapid Coprecipitation Technique Using Yttrium Hydroxide for the Preconcentration and Separation of Trace Elements in Saline Water Prior to Their ICP-AES Determination Shigehiro KAGAYA,† Satoshi MIWA, Toshiyuki MIZUNO, and Koji TOHDA Graduate School of Science and Engineering for Research, University of Toyama, Gofuku 3190, Toyama 930–8555, Japan Yttrium hydroxide quantitatively coprecipitated Be(II), Ti(IV), Cr(III), Mn(II), Fe(III), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), and Pb(II) at pH 9.6 – 10.0 for seawater and pH 10.5 – 11.4 for a table-salt solution. The coprecipitated elements could be determined by inductively coupled plasma atomic emission spectrometry; yttrium was used as an internal standard element. The detection limits ranged from 0.0016 μg (Mn(II)) to 0.22 μg (Zn(II)) in 100 mL of sample solutions. The operation time required to separate 11 elements was approximately 30 min. (Received May 25, 2007; Accepted June 18, 2007; Published August 10, 2007) It is important to determine trace elements in environmental of many elements. However, to our knowledge, no application samples from the viewpoint of environmental management and/or of yttrium hydroxide to the preconcentration/separation of trace protection from hazardous infection1 as well as comprehension elements in environmental water samples has been reported. of the distribution of trace elements in the environment.2 In this work, we developed a preconcentration/separation Recently, multielement profiling analysis, in which major-to- method based on a rapid coprecipitation technique using yttrium ultratrace elements are determined and the distribution patterns hydroxide as a coprecipitant for the determination of trace of the elements are analyzed, has gathered attention to provide elements in environmental water samples, especially saline information about the elemental cycle in the environment.3,4 water. -

Evaluation of Anti-Corrosion and Anti-Galling Performance
EVALUATION OF ANTI-CORROSION AND ANTI-GALLING PERFORMANCE OF A NOVEL GREASE COMPOUND A Thesis by JOHN OLUMIDE REIS Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Hong Liang Committee Members, Chii-Der Suh Sevan Goenezan Head of Department, Andreas A. Polycarpou December 2017 Major Subject: Mechanical Engineering Copyright 2017 John Olumide Reis ABSTRACT In this research, the effectiveness of CeO2, Y2O3 and Al2O3 as anti-corrosion and anti-wear additives in commercial grease was investigated. An experimental approach was used to carry on the research. The weight loss of steel coupons protected with a layer of thin grease, with and without the anti-corrosion additives in a corrosive environment was determined. The Friction factor of the grease compound was also evaluated. The accelerated corrosion tests were performed in a salt spray chamber for an exposure time of 2 weeks (336 hours). The corrosive medium was 5 % wt. of Brine. By varying the weight compositions of the additives (1% wt. and 3% wt.), and comparing the corrosion rates with that of base grease, the effectiveness of various grease additives was evaluated. The result showed that corrosive losses of the test samples can be effectively reduced by adding relatively small amounts of CeO2, Y2O3 and Al2O3 to the base grease. Corroded surfaces were examined using the Optical microscope to clarify the corrosion mechanism. The friction test was carried out using a galling tester. The standard test procedure as specified in API RP 7A1 was followed. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub
US 20050044778A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub. Date: Mar. 3, 2005 (54) FUEL COMPOSITIONS EMPLOYING Publication Classification CATALYST COMBUSTION STRUCTURE (51) Int. CI.' ........ C10L 1/28; C1OL 1/24; C1OL 1/18; (76) Inventor: William C. Orr, Denver, CO (US) C1OL 1/12; C1OL 1/26 Correspondence Address: (52) U.S. Cl. ................. 44/320; 44/435; 44/378; 44/388; HOGAN & HARTSON LLP 44/385; 44/444; 44/443 ONE TABOR CENTER, SUITE 1500 1200 SEVENTEENTH ST DENVER, CO 80202 (US) (57) ABSTRACT (21) Appl. No.: 10/722,127 Metallic vapor phase fuel compositions relating to a broad (22) Filed: Nov. 24, 2003 Spectrum of pollution reducing, improved combustion per Related U.S. Application Data formance, and enhanced Stability fuel compositions for use in jet, aviation, turbine, diesel, gasoline, and other combus (63) Continuation-in-part of application No. 08/986,891, tion applications include co-combustion agents preferably filed on Dec. 8, 1997, now Pat. No. 6,652,608. including trimethoxymethylsilane. Patent Application Publication Mar. 3, 2005 US 2005/0044778A1 FIGURE 1 CALCULATING BUNSEN BURNER LAMINAR FLAME VELOCITY (LFV) OR BURNING VELOCITY (BV) CONVENTIONAL FLAME LUMINOUS FLAME Method For Calculating Bunsen Burner Laminar Flame Velocity (LHV) or Burning Velocity Requires Inside Laminar Cone Angle (0) and The Gas Velocity (Vg). LFV = A, SIN 2 x VG US 2005/0044778A1 Mar. 3, 2005 FUEL COMPOSITIONS EMPLOYING CATALYST Chart of Elements (CAS version), and mixture, wherein said COMBUSTION STRUCTURE element or derivative compound, is combustible, and option 0001) The present invention is a CIP of my U.S. -

Download Download
— Studies on Lithium Acetylide Kenneth N. Campbell and Barbara K. Campbell, The University of Notre Dame In contrast to the large amount of work done on the acetylene derivatives of sodium, potassium and calcium, little attention has been paid to the analogous compounds of lithium. In 1898 Moissani prepared lithium acetylide on a small scale, by the action of acetylene on a liquid ammonia solution of lithium. He reported that lithium acetylide was less soluble in liquid ammonia than sodium acetylide, and that when isolated from the solvent, it was less stable, undergoing decomposition with evolu- tion of acetylene. On the basis of the weight of lithium acetylide obtained from a given weight of lithium, and from the amount of acetylene liberated on hydrolysis, he assigned to lithium acetylide the formula C2Li2.C2H2.2NH3. Since that time no references to lithium acetylide or lithium alkylacetylides have appeared in the literature. It was the pur- pose of the present work, therefore, to prepare and analyze lithium acetylide and a lithium alkylacetylide, and to compare their reactions with those of the better known sodium derivatives. Experimental Procedure Preparation of Lithium and Sodium Acetylides.—Acetylene gas, washed by bubbling through concentrated sulfuric acid, was passed into two liters of liquid ammonia, while 7 g. (1 mole) of metallic lithium, cut in small pieces, was added gradually, with stirring, at a rate such that the solution did not develop a permanent deep blue color. When the solution became colorless after the addition of the last piece of lithium, the flow of acetylene was stopped. -

Experimental Aspects of Geoneutrino Detection: Status and Perspectives
Experimental Aspects of Geoneutrino Detection: Status and Perspectives O. Smirnov,1 1JINR, Joint Institute for Nuclear Research, Dubna, Russian Federation October 22, 2019 Abstract Neutrino geophysics, the study of the Earth's interior by measuring the fluxes of geologically produced neutrino at its surface, is a new interdisciplinary field of science, rapidly developing as a synergy between geology, geophysics and particle physics. Geoneutrinos, antineutrinos from long- lived natural isotopes responsible for the radiogenic heat flux, provide valuable information for the chemical composition models of the Earth. The calculations of the expected geoneutrino signal are discussed, together with experimental aspects of geoneutrino detection, including the description of possible backgrounds and methods for their suppression. At present, only two detectors, Borexino and KamLAND, have reached sensitivity to the geoneutrino. The experiments accumulated a set of ∼190 geoneutrino events and continue the data acquisition. The detailed description of the experiments, their results on geoneutrino detection, and impact on geophysics are presented. The start of operation of other detectors sensitive to geoneutrinos is planned for the near future: the SNO+ detector is being filled with liquid scintillator, and the biggest ever 20 kt JUNO detector is under construction. A review of the physics potential of these experiments with respect to the geoneutrino studies, along with other proposals, is presented. New ideas and methods for geoneutrino detection are reviewed. Contents 1 Introduction 2 2 Geoneutrinos and the Earth's heat 4 2.1 Long-lived radiogenic elements . 4 2.2 Radiogenic heat and geoneutrino luminosity of the Earth . 9 arXiv:1910.09321v1 [physics.geo-ph] 21 Oct 2019 3 Geoneutrino flux calculation 11 3.1 Neutrino oscillations . -

Safety Data Sheet
SAFETY DATA SHEET Preparation Date: 7/21/2017 Revision Date: 7/21/2017 Revision Number: G1 1. IDENTIFICATION Product identifier Product code: S1725 Product Name: SULFUR, LUMP Other means of identification Synonyms: No information available CAS #: 7704-34-9 RTECS # WS4250000 CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: In manufacturing sulfuric acid, carbon disulfide, sulfites, insecticides, plastics, enamels, metal-glass, cements; in vulcanizing rubber; in syntheses of dyes; in making gunpowder, matches; for bleaching wood pulp, straw, wool, silk, felt, linen; in making phosphatic fertilizers; bleaching of dried fruits; fungicide and acaricide; rodent repellent; soil conditioner; nucleating reagent for photographic film; used in cosmetics, such as acne ointments and lotions, and in antidandruff shampoos. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000. Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is considered hazardous according to the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Considered a dangerous substance or mixture according to the Globally Harmonized System (GHS) Flammable solids Category 2 Label elements Warning Flammable solids Product code: S1725 Product name: SULFUR, LUMP 1 / 14 Hazards not otherwise classified (HNOC) Not Applicable Other hazards Not available Precautionary Statements - Prevention Keep away from heat/sparks/open flames/hot surfaces. — No smoking Ground/bond container and receiving equipment Use explosion-proof electrical/ventilating/lighting/.../equipment Wear protective gloves Wear eye/face protection Prevent dust accumulations to minimize explosion hazard In case of fire: Use CO2, dry chemical, or foam to extinguish. -

Chemical Matrix
Chemical Matrix - Ceramics Studio Recommended Personal Protective Key Hazardous Properties Storage Location Disposal Equipment Face Body Hands Protection Respiratory Respiratory Product Form / Phase Key Composition Combustible High Toxicity High Flash Point F Flash Point Plaster Room Plaster Sanitary Drain Sanitary Compressed Gas Compressed Strong Acid/Base Strong Non-Haz Disposal Non-Haz Non-Flam Cabinet Non-Flam Glaze Mixing Room Haz Waste Disposal Waste Haz Flammables Cabinet Flammable/Explosive Potentially SensitizingPotentially MSDS Strong Oxidizer / Reducer Oxidizer Strong Cartirdge Face Shield Nitrile Gloves Nitrile Use with Local Only Exhaust Ventilation with Use Non-Flam Cupboard / Shelving / Counter Cupboard Non-Flam Chemical Splash Apron Chemical Safety Glasses Safety Chemical Flame Resistant Lab Coat Lab Resistant Flame Half Face Respitator with P100 P100 with Respitator Face Half Chemical Protective Gloves per per Gloves Protective Chemical Acetone Liquid Acetone -4 X X X X X X X Albany Slip Powder Silty glacial clay used for pottery, contains up to 30% quartz free silica according to Columbus Clay web X X X X X X MSDS Alumina Hydrate Powder Alumina Hydrate X X X X X X Angelos Copperslip Liquid (thick) Copper oxide, frit 3110, CMC gum, bentonite X X Antimony oxide Powder Antimony oxide X X X X X X X X Baking Soda Powder Sodium bicarbonate X X X X X X X Bell Dark Ball Clay Powder Kaolinite, plus up to 30% quartz free silica X X X X X X Bernard Slip Clay Powder Clay, plus up to 30% silica, some considered respirable according -
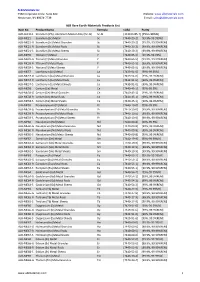
ALB Materials Inc Product List.Xlsx
ALB Materials Inc 2360 Corporate Circle. Suite 400 Website: www.albmaterials.com Henderson, NV 89074-7739 E-mail: [email protected] ALB Rare Earth Materials Products List Item No. Product Name Formula CAS# Purity ALB-AL2113 Scandium (2%)- Aluminum Master Alloy (Sc-Al) Sc-Al [113413-85-7] [2%Sc+98%Al] ALB-ME21 Scandium (Sc) Metal Sc [7440-20-2] [99.9%-99.999%] ALB-ME21-G Scandium (Sc) Metal Granules Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-R Scandium (Sc) Metal Rods Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-S Scandium (Sc) Metal Sheets Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME39 Yttrium (Y) Metal Y [7440-65-5] [99.9%-99.99%] ALB-ME39-G Yttrium (Y) Metal Granules Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-R Yttrium (Y) Metal Rods Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-S Yttrium (Y) Metal Sheets Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME57 Lanthanum (La) Metal La [7439-91-0] [99%-99.95%] ALB-ME57-G Lanthanum (La) Metal Granules La [7439-91-0] [99%, 99.9%REM] ALB-ME57-R Lanthanum (La) Metal Rods La [7439-91-0] [99%, 99.9%REM] ALB-ME57-S Lanthanum (La) Metal Sheets La [7439-91-0] [99%, 99.9%REM] ALB-ME58 Cerium (Ce) Metal Ce [7440-45-1] [99%-99.9%] ALB-ME58-G Cerium (Ce) Metal Granules Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-R Cerium (Ce) Metal Rods Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-S Cerium (Ce) Metal Sheets Ce [7440-45-1] [99%, 99.9%REM] ALB-ME59 Praseodymium (Pr) Metal Pr [7440-10-0] [99%-99.9%] ALB-ME59-G Praseodymium (Pr) Metal Granules Pr [7440-10-0] [99.9%, 99.99%REM] ALB-ME59-R Praseodymium (Pr) Metal Rods Pr [7440-10-0] -

The Action of Nitrogen on Lithium Carbide. by S
‘478 GENERAL, PHYSICAL AND INORGANIC. chain action as suggested by Daneel,’ then anything which alters the degree of association of the water would influence the magnitude of this effect. In order, therefore, to estimate the degree of ionization of strong acids or alkalies in concentrated solutions, the best procedure would probably be to make use of the equation 7 = AjAb (34) in which A is the equivalent conductance of the solution for which r is desired, and above which the vapor pressure of the water is p. A’, is the equivalent conductance of fie electrolyte at infinit dilution in a solu- tion to which a suitable non-electrolyte has been added, so as to give it the same viscosity and the same vapor pressure, p, as the first solution. In the absence of any experimental data, however, equation (34) must be regarded merely as a suggestion. Summary. I. It is pointed out that in calculating the degree of ionization of an electrolyte by the conductivity method, the neglect of the viscosity effect may produce errors as high as 7 and 8 per cent., even at such low concentrations as 0.1 normal and for such simple electrolytes as uni- univalent salts. 2. The relation A = kj” (in which A is the equivalent conductance of an ion, f is the fluidity of the solution, m is a constant not far from unity and dependent chiefly upon the nature of the ion, and k is a propor- tionality constant) is proposed, provisionally as the most logical basis for formulating the viscosity correction in calculating the degree of ioniza- tion, y, in moderately concentrated solutions. -

Zirconia-Ceria-Yttria-Based Mixed Oxide and Process for Producing The
(19) TZZ_ _Z_T (11) EP 1 921 044 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C01G 25/00 (2006.01) B01J 23/10 (2006.01) 28.02.2018 Bulletin 2018/09 B01D 53/94 (2006.01) B01J 21/06 (2006.01) B01J 35/10 (2006.01) (21) Application number: 07118246.3 (22) Date of filing: 10.10.2007 (54) Zirconia-ceria-yttria-based mixed oxide and process for producing the same Auf Zirkonium-Cerium-Yttrium basierendes Mischoxid und Herstellungsverfahren dafür Oxyde mixte à base de zirconium-cérium-yttrium et son procédé de fabrication (84) Designated Contracting States: • Kodama, Hiroshi DE FR GB Osaka 5590025 (JP) (30) Priority: 12.10.2006 JP 2006305934 (74) Representative: Manley, Nicholas Michael 08.08.2007 JP 2007206394 WP Thompson 8th Floor (43) Date of publication of application: 1 Mann Island 14.05.2008 Bulletin 2008/20 Liverpool L3 1BP (GB) (73) Proprietor: Daiichi Kigenso Kagaku Kogyo Co., (56) References cited: Ltd. EP-A- 1 006 081 EP-A- 1 035 074 Osaka-shi, EP-A- 1 894 620 WO-A-2007/093593 Osaka 559-0025 (JP) JP-A- 2003 277 059 US-B1- 6 387 338 (72) Inventors: • Okamoto, Hiroshi Osaka 5590025 (JP) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been paid. -

Depositional Characteristics of Recent and Late Holocene Overwash Sandsheets in Coastal Embayments from Southeast Australia
Depositional characteristics of recent and late Holocene overwash sandsheets in coastal embayments from southeast Australia. Adam D Switzer BSc (Hons) A thesis submitted in part fulfilment of the requirements of the Doctor of Philosophy in the School of Earth and Environmental Sciences University of Wollongong February 2005 CERTIFICATION I, Adam D. Switzer, declare that this thesis, submitted in partial fulfilment of the requirements for the award of Doctor of Philosophy, in the School of earth and Environmental Sciences, University of Wollongong, is wholly my own work unless otherwise referenced or acknowledged. The document has not been submitted for qualifications at any other academic institution. Adam D. Switzer 16 February 2005 ii Acknowledgments This project has benefited considerably from the leadership and input of two very unique and different individuals, my academic supervisors Brian Jones and Ted Bryant. Working with them has provided an inspirational, fascinating and sometimes frustrating insight into the world of academic studies. This project also benefited considerably from the academic input of Charlie Bristow, Simon Haslett, Colin Woodroffe, Allan Chivas and Bryan Chenhall. A major part of the funding for this project comes from Dunmore Sand and Soil and I would sincerely like to thank Kerry Steggles, Managing Director for his support of the project. The other major funding component for the project came from an Australian Postgraduate Award (Industry) and I am greatly appreciative of that. Like many people who undertake a major project I have often needed to draw on the support of my family and friends. Naomi Riggs is the one person who heads up this team.