The Thermodynamic Properties of Neodymium Hydroxide In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Improvement of High−Temperature Characteristics of the Sintered
686 EJ60ケooho勿zゑsψりノ 一Article一 Improvement of High-Temperature Characteristics of the Sintered Nickel Positive Electrode for an Alkaline Storage Battery Katsuhiko SHINYAMA,*Yoshifumi MAGARI,Atsuhiro FUNAHASHI, Toshiyuki NOHMA,and Ikuo YONEZU R&D Business Unit,Mobile Energy Company,Sanyo Electric Co.,Ltd.(7-3-21bukidai-higashimachi,Nishi-ku,Kobe City,Hyogo651-2242,Japan) Received August9,2002;Accepted May20,2003 The high-temperature characteristics of sintered nickel positive electrodes for alkaline storage batteries such as nickel・metal hydride batteries and nicke1-cadmium batteries were investigated.Generally,the discharge capacity of an alkaline storage battery charged at high temperature is smaller than that charged at room temperature due to the oxygen evolution reaction.In order to enhance high-temperature characteristics,we coated sintered n量ckel positive electrodes with yttrium hydroxide,calcium hydroxide or cobalt hydroxide.Th6high-temperature characteristics of the sintered nickel positive electrodes coated with y血ium hydroxide and calcium hydroxide by immersing them in sodium hydroxide solution after immersing in nitrate solution were greatly enhanced because of the increased oxy. gen OVerVOltage. κ¢y防鳩:Sintered Nickel Positive Electrode,High-temperature Characteristics,Alkaline Storage Battery,Oxygen Overvoltage l lntroduction In the past,the electrolyte composition3)and dissolv- Cun℃ntly,alkaline storage batteries such as nicke1- ing such elements as cobalt and calcium into nickel hy- cadmium batteries and nickel-metal hydride -

Rapid Coprecipitation Technique Using Yttrium Hydroxide for the Preconcentration and Separation of Trace Elements in Saline Water Prior to Their ICP-AES Determination
ANALYTICAL SCIENCES AUGUST 2007, VOL. 23 1021 2007 © The Japan Society for Analytical Chemistry Notes Rapid Coprecipitation Technique Using Yttrium Hydroxide for the Preconcentration and Separation of Trace Elements in Saline Water Prior to Their ICP-AES Determination Shigehiro KAGAYA,† Satoshi MIWA, Toshiyuki MIZUNO, and Koji TOHDA Graduate School of Science and Engineering for Research, University of Toyama, Gofuku 3190, Toyama 930–8555, Japan Yttrium hydroxide quantitatively coprecipitated Be(II), Ti(IV), Cr(III), Mn(II), Fe(III), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), and Pb(II) at pH 9.6 – 10.0 for seawater and pH 10.5 – 11.4 for a table-salt solution. The coprecipitated elements could be determined by inductively coupled plasma atomic emission spectrometry; yttrium was used as an internal standard element. The detection limits ranged from 0.0016 μg (Mn(II)) to 0.22 μg (Zn(II)) in 100 mL of sample solutions. The operation time required to separate 11 elements was approximately 30 min. (Received May 25, 2007; Accepted June 18, 2007; Published August 10, 2007) It is important to determine trace elements in environmental of many elements. However, to our knowledge, no application samples from the viewpoint of environmental management and/or of yttrium hydroxide to the preconcentration/separation of trace protection from hazardous infection1 as well as comprehension elements in environmental water samples has been reported. of the distribution of trace elements in the environment.2 In this work, we developed a preconcentration/separation Recently, multielement profiling analysis, in which major-to- method based on a rapid coprecipitation technique using yttrium ultratrace elements are determined and the distribution patterns hydroxide as a coprecipitant for the determination of trace of the elements are analyzed, has gathered attention to provide elements in environmental water samples, especially saline information about the elemental cycle in the environment.3,4 water. -

Evaluation of Anti-Corrosion and Anti-Galling Performance
EVALUATION OF ANTI-CORROSION AND ANTI-GALLING PERFORMANCE OF A NOVEL GREASE COMPOUND A Thesis by JOHN OLUMIDE REIS Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Hong Liang Committee Members, Chii-Der Suh Sevan Goenezan Head of Department, Andreas A. Polycarpou December 2017 Major Subject: Mechanical Engineering Copyright 2017 John Olumide Reis ABSTRACT In this research, the effectiveness of CeO2, Y2O3 and Al2O3 as anti-corrosion and anti-wear additives in commercial grease was investigated. An experimental approach was used to carry on the research. The weight loss of steel coupons protected with a layer of thin grease, with and without the anti-corrosion additives in a corrosive environment was determined. The Friction factor of the grease compound was also evaluated. The accelerated corrosion tests were performed in a salt spray chamber for an exposure time of 2 weeks (336 hours). The corrosive medium was 5 % wt. of Brine. By varying the weight compositions of the additives (1% wt. and 3% wt.), and comparing the corrosion rates with that of base grease, the effectiveness of various grease additives was evaluated. The result showed that corrosive losses of the test samples can be effectively reduced by adding relatively small amounts of CeO2, Y2O3 and Al2O3 to the base grease. Corroded surfaces were examined using the Optical microscope to clarify the corrosion mechanism. The friction test was carried out using a galling tester. The standard test procedure as specified in API RP 7A1 was followed. -
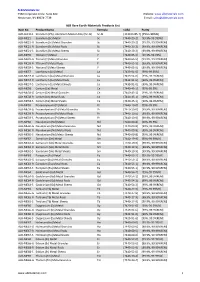
ALB Materials Inc Product List.Xlsx
ALB Materials Inc 2360 Corporate Circle. Suite 400 Website: www.albmaterials.com Henderson, NV 89074-7739 E-mail: [email protected] ALB Rare Earth Materials Products List Item No. Product Name Formula CAS# Purity ALB-AL2113 Scandium (2%)- Aluminum Master Alloy (Sc-Al) Sc-Al [113413-85-7] [2%Sc+98%Al] ALB-ME21 Scandium (Sc) Metal Sc [7440-20-2] [99.9%-99.999%] ALB-ME21-G Scandium (Sc) Metal Granules Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-R Scandium (Sc) Metal Rods Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME21-S Scandium (Sc) Metal Sheets Sc [7440-20-2] [99.9%, 99.99%REM] ALB-ME39 Yttrium (Y) Metal Y [7440-65-5] [99.9%-99.99%] ALB-ME39-G Yttrium (Y) Metal Granules Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-R Yttrium (Y) Metal Rods Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME39-S Yttrium (Y) Metal Sheets Y [7440-65-5] [99.9%, 99.99%REM] ALB-ME57 Lanthanum (La) Metal La [7439-91-0] [99%-99.95%] ALB-ME57-G Lanthanum (La) Metal Granules La [7439-91-0] [99%, 99.9%REM] ALB-ME57-R Lanthanum (La) Metal Rods La [7439-91-0] [99%, 99.9%REM] ALB-ME57-S Lanthanum (La) Metal Sheets La [7439-91-0] [99%, 99.9%REM] ALB-ME58 Cerium (Ce) Metal Ce [7440-45-1] [99%-99.9%] ALB-ME58-G Cerium (Ce) Metal Granules Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-R Cerium (Ce) Metal Rods Ce [7440-45-1] [99%, 99.9%REM] ALB-ME58-S Cerium (Ce) Metal Sheets Ce [7440-45-1] [99%, 99.9%REM] ALB-ME59 Praseodymium (Pr) Metal Pr [7440-10-0] [99%-99.9%] ALB-ME59-G Praseodymium (Pr) Metal Granules Pr [7440-10-0] [99.9%, 99.99%REM] ALB-ME59-R Praseodymium (Pr) Metal Rods Pr [7440-10-0] -

Zirconia-Ceria-Yttria-Based Mixed Oxide and Process for Producing The
(19) TZZ_ _Z_T (11) EP 1 921 044 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C01G 25/00 (2006.01) B01J 23/10 (2006.01) 28.02.2018 Bulletin 2018/09 B01D 53/94 (2006.01) B01J 21/06 (2006.01) B01J 35/10 (2006.01) (21) Application number: 07118246.3 (22) Date of filing: 10.10.2007 (54) Zirconia-ceria-yttria-based mixed oxide and process for producing the same Auf Zirkonium-Cerium-Yttrium basierendes Mischoxid und Herstellungsverfahren dafür Oxyde mixte à base de zirconium-cérium-yttrium et son procédé de fabrication (84) Designated Contracting States: • Kodama, Hiroshi DE FR GB Osaka 5590025 (JP) (30) Priority: 12.10.2006 JP 2006305934 (74) Representative: Manley, Nicholas Michael 08.08.2007 JP 2007206394 WP Thompson 8th Floor (43) Date of publication of application: 1 Mann Island 14.05.2008 Bulletin 2008/20 Liverpool L3 1BP (GB) (73) Proprietor: Daiichi Kigenso Kagaku Kogyo Co., (56) References cited: Ltd. EP-A- 1 006 081 EP-A- 1 035 074 Osaka-shi, EP-A- 1 894 620 WO-A-2007/093593 Osaka 559-0025 (JP) JP-A- 2003 277 059 US-B1- 6 387 338 (72) Inventors: • Okamoto, Hiroshi Osaka 5590025 (JP) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been paid. -

Study on Extraction of Lanthanum Oxide from Monazite Concentrate
World Academy of Science, Engineering and Technology International Journal of Materials and Metallurgical Engineering Vol:2, No:10, 2008 Study on Extraction of Lanthanum Oxide from Monazite Concentrate Nwe Nwe Soe, Lwin Thuzar Shwe, and Kay Thi Lwin La2O3 has more extensive applications. It is a white Abstract—Lanthanum oxide is to be recovered from monazite, amorphous powder; insoluble in water improves the alkali which contains about 13.44% lanthanum oxide. The principal resistance of glass. The Kodak's camera objectives contain objective of this study is to be able to extract lanthanum oxide from from 20 to 40% La2O3. Lanthanum oxide can also be used in monazite of Moemeik Myitsone Area. The treatment of monazite in optical glasses where it imparts improved alkali resistance, this study involves three main steps; extraction of lanthanum La-Ce-Tb phosphors for fluorescent lamps, dielectric and hydroxide from monazite by using caustic soda, digestion with nitric conductive ceramics, Barium titanate capacitors, X-Ray acid and precipitation with ammonium hydroxide and calcination of lanthanum oxalate to lanthanum oxide. intensifying screens and lanthanum metal production [1]. Keywords—Calcination, Digestion, Precipitation. II. EXPERIMENTAL PROCEDURE Monazite from Moemeik Myitsone Area was used as raw I. INTRODUCTION material for the experiments. Monazite was ground to obtain - ANTHANUM was discovered in 1839 by Swedish 325 mesh. It has beach color. The flow diagram of the L Chemist Carl Gustav Mosandar when he partially extraction of lanthanum oxide is shown in Fig. 1. decomposed a sample of cerium nitrate by heating and treating Monazite the resulting salt with nitric acid. -

United States Patent [151 3,642,527 Purdes Et Al
United States Patent [151 3,642,527 Purdes et al. v [45] Feb. 15, 1972 [54] METHOD OF MODIFYING va dielectric substrate, such as barium titanate, may be ELECTRICAL RESISTIVITY modi?ed by ?rst forming a relatively porous substrate which CHARACTERISTICS OF DIELECTRIC may be handled without breaking, as by pre?ring the sub SUBSTRATES strate, masking selected portions of the substrate with a material such as a photoresist material which will vaporize [72] Inventors: Andrew J. Purdes, Pawtucket, R.I.; Ernest during ?nal ?ring of the substrate, contacting the substrate ' M. Just, Plainville, Mass. with a solution of a ?rst reactant, immersing at least a portion [ 73] Assignee: Texas Instruments Incorporated, Dallas, of the substrate in a solution of a second reactant which will Tex. react with the ?rst reactant to precipitate in situ in a portion of the substrate a compound which is insoluble in the solutions [22] Filed: Dec. 30, 1968 and which is adapted to modify the electrical resistivity characteristics of the substrate, and thereafter ?ring the sub [21] Appl. No.: 787,989 strate at a temperature on the order of l,400°-l,450° C. to reduce the porosity of the substrate and to incorporate the in {52] us. c1 ....................................... ..117/212, 117/223 soluble compound into the lattice of selected portions of the [511 1m. (:1 ...................................... ..B44d l/l8 substrate. Where it is desired to dope selected portions of an [58] Field 6: Search .................. ..106/39; 117/212, 213, 123, undoped substrate to the desired thickness and form thick ?lm positive temperature coef?cient (PTC) thermistors, the ‘ 117/223 starting material may be an undoped barium titanate, for ex~ [56] References Cited ample, the solution of the ?rst reactant may be an aqueous solution of a compound such as ammonium hydroxide, and UNITED STATES PATENTS the ‘solution of the second reactant may be an aqueous solu tion of a compound such as lanthanum acetate which reacts 3,296,359 1/1967 Ramsey, Jr. -

Tompbnents of Fission and 'Natural Radioactivity Found in Water Are
DOCUMENT RESUME,, A ED '170 1110 SE027 bill. 0 AUTHOR Thatcher, L.L. ;And Others 1 TITLE Techniques of Water-Resources Investigations of the United. States' Geblogical.Survey. Book 5, Laboratory, Analysis. Chapter- A5, Methodscr Dete-rmination of - Radioactive Substances in Water and Fluvial Sediments. INSTITUTION -Geological Survey (Dept,: .of Interior) ,Reston, PUB DATE 77 NO Tr 95p. ;For related document, see ED 131 218 AVAILABLE FRQM Super.intendent of Documents, U.S. Government Printing Off /ce , Washington, D.C. -20402(Stock Number 024- 0.01- 02928- 6:, No price quoted) */ FDRS PRICE MF01/PC04 Plus Postage. DESCRIPTORS *ChemiCal Analysis; *Geolo'gy; *LaboratorTechniques; PhYsic; Pollution; Retource Materials; S.c ence Equipment; Water Pollution control IDENTIFIERS *RadioactiveVs- Substances; *Sediments; Water Quality ABSTkACT Analytical methods for determining important , tompbnents of fission and 'natural radioactivity found in water are reportedThe discussion of each method includes conditions for application of the method, a summary of the method, interferences, required apparatus, Procedures, calculations and estimation of precision. Isotopes considered are cesium-137, strontium-90, lead-210, radium-226. and 228, tritium, and carbon-714. `When two methods are in use for an isotope,- both methods are reported. Techniques for the collection and preservation of water samples to be anallze?d for radioactivity are also discuSsed.(CS) v. *********************************************************************** 4 Re. productions supplied -

Interagency Committee on Chemical Management
DECEMBER 14, 2018 INTERAGENCY COMMITTEE ON CHEMICAL MANAGEMENT EXECUTIVE ORDER NO. 13-17 REPORT TO THE GOVERNOR WALKE, PETER Table of Contents Executive Summary ...................................................................................................................... 2 I. Introduction .......................................................................................................................... 3 II. Recommended Statutory Amendments or Regulatory Changes to Existing Recordkeeping and Reporting Requirements that are Required to Facilitate Assessment of Risks to Human Health and the Environment Posed by Chemical Use in the State ............................................................................................................................ 5 III. Summary of Chemical Use in the State Based on Reported Chemical Inventories....... 8 IV. Summary of Identified Risks to Human Health and the Environment from Reported Chemical Inventories ........................................................................................................... 9 V. Summary of any change under Federal Statute or Rule affecting the Regulation of Chemicals in the State ....................................................................................................... 12 VI. Recommended Legislative or Regulatory Action to Reduce Risks to Human Health and the Environment from Regulated and Unregulated Chemicals of Emerging Concern .............................................................................................................................. -

Coprecipitation of Yttrium and Aluminium Hydroxide for Preparation of Yttrium Aluminium Garnet
Coprecipitation of yttrium and aluminium hydroxide for preparation of yttrium aluminium garnet Citation for published version (APA): Vrolijk, J. W. G. A., Willems, J. W. M. M., & Metselaar, R. (1990). Coprecipitation of yttrium and aluminium hydroxide for preparation of yttrium aluminium garnet. Journal of the European Ceramic Society, 6(1), 47-51. https://doi.org/10.1016/0955-2219(90)90034-D DOI: 10.1016/0955-2219(90)90034-D Document status and date: Published: 01/01/1990 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. -

Hydrophilic Block Copolymer-Directed Growth of Lanthanum Hydroxide Nanoparticles
PAPER www.rsc.org/njc | New Journal of Chemistry Hydrophilic block copolymer-directed growth of lanthanum hydroxide nanoparticles Fre´de´ric Bouyer,a Nicolas Sanson,a Mathias Destaracb and Corine Ge´rardin*a Received (in Montpellier, France) 18th November 2005, Accepted 21st December 2005 First published as an Advance Article on the web 25th January 2006 DOI: 10.1039/b516368d Stable hairy lanthanum hydroxide nanoparticles were synthesized in water by performing hydrolysis and condensation reactions of lanthanum cations in the presence of double hydrophilic polyacrylic acid-b-polyacrylamide block copolymers (PAA-b-PAM). In the first step, the addition of asymmetric PAA-b-PAM copolymers (Mw,PAA o Mw,PAM) to lanthanum salt solutions, both at pH = 5.5, induces the formation of monodispersed micellar aggregates, which are predominantly isotropic. The core of the hybrid aggregates is constituted of a lanthanum polyacrylate complex whose formation is due to bidentate coordination bonding between La31 and acrylate groups, as shown by ATR-FTIR experiments and pH measurements. The size of the micellar aggregates depends on the molecular weight of the copolymer but is independent of the copolymer to metal ratio in solution. In the second step, the hydrolysis of lanthanum ions is induced by addition of a strong base such as sodium hydroxide. Either flocculated suspensions or stable anisotropic or spherical nanoparticles of lanthanum hydrolysis products were obtained depending on the metal complexation ratio [acrylate]/[La]. The variation of that parameter also enables the control of the size of the core-corona nanoparticles obtained by lanthanum hydroxylation. The asymmetry degree of the copolymer was shown to influence both the size and the shape of the particles. -

Arsenic Treatment Technologies for Soil, Waste, and Water
Appendix A Literature Search Results Dialog® References Citation Reviewed Removal of arsenic from drinking water using modified fly-ash bed Goswami, D.; Das, A.K. Int. J. Water, Vol. 1, No. 1, pp. 61-70 (2000) DOCUMENT TYPE: Journal LANGUAGE: ENGLISH Ferrous iron treatment of soils contaminated with arsenic-containing T wood-preserving solution Moore, T.J.; Rightmire, C.M.; Vempati, R.K. Soil Sediment Contam., Vol. 9, No. 4, pp. 375-405 (2000) DOCUMENT TYPE: Journal LANGUAGE: ENGLISH Development of a sulfate-reducing biological process to remove heavy metals from acid mine drainage Steed, V.S.; Suidan, M.T.; Gupta, M.; Miyahara, T.; Acheson, C.M.; Sayles, G.D. Water Environ. Res., Vol. 72, No. 5, pp. 530-535 (Sep.-Oct. 2000) DOCUMENT TYPE: Journal LANGUAGE: ENGLISH Metal behavior during the low-temperature pyrolysis of chromated copper arsenate-treated wood waste Helsen, L.; Van den Bulck, E. Environ. Sci. Technol., Vol. 34, No. 14, pp. 2931-2938 (15 Jul. DOCUMENT TYPE: Journal LANGUAGE: ENGLISH Biochemical processes for geothermal brine treatment Premuzic, E.T.; Lin, M.S.; Bohenek, M. et al Geothermal: the Clean & Green Energy Choice for the World. Geothermal Resources Council, (1998). Transactions of the Geothermal Resources Council No. 22. pp. 441-444 DOCUMENT TYPE: Book LANGUAGE: ENGLISH Management of arsenic wastes: problems and prospects Leist,M; Casey, R.J; Caridi, D; J. Hazard Mater., Vol 76, No. 1, pp. 125-138 (28 Aug 2000) DOCUMENT TYPE: Journal LANGUAGE: ENGLISH Treatment of arsenic-contaminated soils. II: treatability study and T remediation Miller, J.; Akhter, H.; Cartledge, F.K.: McLearn, M J.