Downloaded for Personal Non-Commercial Research Or Study, Without Prior Permission Or Charge
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interaction of Shikimic Acid with Shikimate Kinase
BBRC Biochemical and Biophysical Research Communications 325 (2004) 10–17 www.elsevier.com/locate/ybbrc Interaction of shikimic acid with shikimate kinase Jose´ Henrique Pereiraa, Jaim Simo˜es de Oliveirab, Fernanda Canduria,c, Marcio Vinicius Bertacine Diasa,Ma´rio Se´rgio Palmac,d, Luiz Augusto Bassob, Walter Filgueira de Azevedo Jr.a,d,*, Dio´genes Santiago Santose,* a Department of Physics, UNESP, Sa˜o Jose´ do Rio Preto, SP 15054-000, Brazil b Rede Brasileira de Pesquisa em Tuberculose Grupo de Microbiologia Molecular e Funcional, Departamento de Biologia Molecular e Biotecnologia, UFRGS, Porto Alegre, RS 91501-970, Brazil c Center for Applied Toxinology, Institute Butantan, Sa˜o Paulo, SP 05503-900, Brazil d Laboratory of Structural Biology and Zoochemistry, CEIS/Department of Biology, Institute of Biosciences, UNESP, Rio Claro, SP 13506-900, Brazil e Centro de Pesquisa e Desenvolvimento em Biologia Molecular e Funcional, Pontifı´cia Universidade Cato´lica do Rio Grande do Sul, Porto Alegre, RS 90619-900, Brazil Received 24 September 2004 Available online 19 October 2004 Abstract The crystal structure of shikimate kinase from Mycobacterium tuberculosis (MtSK) complexed with MgADP and shikimic acid (shikimate) has been determined at 2.3 A˚ resolution, clearly revealing the amino acid residues involved in shikimate binding. In MtSK, the Glu61 strictly conserved in SK forms a hydrogen bond and salt-bridge with Arg58 and assists in positioning the guan- idinium group of Arg58 for shikimate binding. The carboxyl group of shikimate interacts with Arg58, Gly81, and Arg136, and hydroxyl groups with Asp34 and Gly80. The crystal structure of MtSK–MgADP–shikimate will provide crucial information for elucidation of the mechanism of SK-catalyzed reaction and for the development of a new generation of drugs against tuberculosis. -

Genome-Scale Fitness Profile of Caulobacter Crescentus Grown in Natural Freshwater
Supplemental Material Genome-scale fitness profile of Caulobacter crescentus grown in natural freshwater Kristy L. Hentchel, Leila M. Reyes Ruiz, Aretha Fiebig, Patrick D. Curtis, Maureen L. Coleman, Sean Crosson Tn5 and Tn-Himar: comparing gene essentiality and the effects of gene disruption on fitness across studies A previous analysis of a highly saturated Caulobacter Tn5 transposon library revealed a set of genes that are required for growth in complex PYE medium [1]; approximately 14% of genes in the genome were deemed essential. The total genome insertion coverage was lower in the Himar library described here than in the Tn5 dataset of Christen et al (2011), as Tn-Himar inserts specifically into TA dinucleotide sites (with 67% GC content, TA sites are relatively limited in the Caulobacter genome). Genes for which we failed to detect Tn-Himar insertions (Table S13) were largely consistent with essential genes reported by Christen et al [1], with exceptions likely due to differential coverage of Tn5 versus Tn-Himar mutagenesis and differences in metrics used to define essentiality. A comparison of the essential genes defined by Christen et al and by our Tn5-seq and Tn-Himar fitness studies is presented in Table S4. We have uncovered evidence for gene disruptions that both enhanced or reduced strain fitness in lake water and M2X relative to PYE. Such results are consistent for a number of genes across both the Tn5 and Tn-Himar datasets. Disruption of genes encoding three metabolic enzymes, a class C β-lactamase family protein (CCNA_00255), transaldolase (CCNA_03729), and methylcrotonyl-CoA carboxylase (CCNA_02250), enhanced Caulobacter fitness in Lake Michigan water relative to PYE using both Tn5 and Tn-Himar approaches (Table S7). -

Identification of Active Molecules Against Mycobacterial Shikimate
Identification of active molecules against Mycobacterial Shikimate Kinase from Chemical library and their affinity with different domains Sapna Pandey1, Ekta Dhamija1, Sanjay Kumar1, Pragya Yadav1, Tadigopula Narender1, Arnava Dasgupta1, Ravishankar Ramachandran1, and KISHORE SRIVASTAVA1 1Central Drug Research Institute November 9, 2020 Abstract Tuberculosis (TB), regardless of being the oldest disease is still a menace that humans have not been able to control. With the advancement in the drug discovery programme, target-based drug discovery appears to be one of the promising techniques for the development of future therapeutics. It involves identifying an essential gene involved in the pathogenesis of the disease and then targeting the protein against a defined chemical library. Shikimate kinase is one such validated target in mycobacterium. It is vital for the growth of bacteria and is absent in mammals, making it an ideal drug target. Here 6427 compounds were screened through structure based virtual screening where compound S-014-1049 was found active against H37Rv and proven non-cytotoxic in in vitro studies. It specifically binds to the core domain of MTSK. Introduction The development of drug resistance is a severe threat to public health. Presently, use of antibiotics targets bacterial structure and essential functions. Due to the well-identified anti-mycobacterial resistance, there has been an increased interest in the discovery of novel drugs to target other essential processes of bacterial survival. Currently, target-based drug screening is promising and efficient way for development of therapeutic agents. In recent years, extensive efforts have been made for the discovery of inhibitors of enzymes involved in the biosynthesis of aromatic amino acids. -

The Role of the Salvage Pathway in Nucleotide Sugar Biosynthesis
THE ROLE OF THE SALVAGE PATHWAY IN NUCLEOTIDE SUGAR BIOSYNTHESIS: IDENTIFICATION OF SUGAR KINASES AND NDP-SUGAR PYROPHOSPHORYLASES by TING YANG (Under the Direction of Maor Bar-Peled) ABSTRACT The synthesis of polysaccharides, glycoproteins, glycolipids, glycosylated secondary metabolites and hormones requires a large number of glycosyltransferases and a constant supply of nucleotide sugars. In plants, photosynthesis and the NDP-sugar inter-conversion pathway are the major entry points to form NDP-sugars. In addition to these pathways is the salvage pathway, a less understood metabolism that provides the flux of NDP-sugars. This latter pathway involves the hydrolysis of glycans to free sugars, sugar transport, sugar phosphorylation and nucleotidylation. The balance between glycan synthesis and recycling as well as its regulation at various plant developmental stages remains elusive as many of the molecular components are unknown. To understand how the salvage pathway contributes to the sugar flux and cell wall biosynthesis, my research focused on the functional identification of salvage pathway sugar kinases and NDP-sugar pyrophosphorylases. This research led to the first identification and enzymatic characterization of galacturonic acid kinase (GalA kinase), galactokinase (GalK), a broad UDP-sugar pyrophosphorylase (sloppy), two promiscuous UDP-GlcNAc pyrophosphorylases (GlcNAc-1-P uridylyltransferases), as well as UDP-sugar pyrophosphorylase paralogs from Trypanosoma cruzi and Leishmania major. To evaluate the salvage pathway in plant biology, we further investigated a sugar kinase mutant: galacturonic acid kinase mutant (galak) and determined if and how galak KO mutant affects the synthesis of glycans in Arabidopsis. Feeding galacturonic acid to the seedlings exhibited a 40-fold accumulation of free GalA in galak mutant, while the wild type (WT) plant readily metabolizes the fed-sugar. -

The Important Structural Role of the P-Loop Lysine
Biochemical and X-ray crystallographic studies on shikimate kinase: The important structural role of the P-loop lysine TINO KRELL,1,6,7 JOHN MACLEAN,2,6,8 DEBORAH J. BOAM,1 ALAN COOPER,2 MARINA RESMINI,3 KEITH BROCKLEHURST,4 SHARON M. KELLY,5 5 2 1 NICHOLAS C. PRICE, ADRIAN J. LAPTHORN, AND JOHN R. COGGINS 1Division of Biochemistry and Molecular Biology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow G12 8QQ, Scotland, UK 2Department of Chemistry, University of Glasgow, Glasgow G12 8QQ, Scotland, UK 3Department of Chemistry, Queen Mary and Westfield College, University of London, London E1 4NS, UK 4Department of Molecular and Cellular Biology, Queen Mary and Westfield College, University of London, London E1 4NS, UK 5Department of Biological Sciences, University of Stirling, Stirling FK9 4 LA, Scotland, UK (RECEIVED December 20, 2000; FINAL REVISION March 8, 2001; ACCEPTED March 12, 2001) Abstract Shikimate kinase, despite low sequence identity, has been shown to be structurally a member of the nucleoside monophosphate (NMP) kinase family, which includes adenylate kinase. In this paper we have explored the roles of residues in the P-loop of shikimate kinase, which forms the binding site for nucleotides and is one of the most conserved structural features in proteins. In common with many members of the P-loop family, shikimate kinase contains a cysteine residue 2 amino acids upstream of the essential lysine residue; the side chains of these residues are shown to form an ion pair. The C13S mutant of shikimate kinase was found to be enzymatically active, whereas the K15M mutant was inactive. -

Conserved Phosphoryl Transfer Mechanisms Within Kinase Families
Kenyon et al. BMC Research Notes 2012, 5:131 http://www.biomedcentral.com/1756-0500/5/131 RESEARCHARTICLE Open Access Conserved phosphoryl transfer mechanisms within kinase families and the role of the C8 proton of ATP in the activation of phosphoryl transfer Colin P Kenyon*, Robyn L Roth, Chris W van der Westhuyzen and Christopher J Parkinson Abstract Background: The kinome is made up of a large number of functionally diverse enzymes, with the classification indicating very little about the extent of the conserved kinetic mechanisms associated with phosphoryl transfer. It has been demonstrated that C8-H of ATP plays a critical role in the activity of a range of kinase and synthetase enzymes. Results: A number of conserved mechanisms within the prescribed kinase fold families have been identified directly utilizing the C8-H of ATP in the initiation of phosphoryl transfer. These mechanisms are based on structurally conserved amino acid residues that are within hydrogen bonding distance of a co-crystallized nucleotide. On the basis of these conserved mechanisms, the role of the nucleotide C8-H in initiating the formation of a pentavalent intermediate between the g-phosphate of the ATP and the substrate nucleophile is defined. All reactions can be clustered into two mechanisms by which the C8-H is induced to be labile via the coordination of a backbone carbonyl to C6-NH2 of the adenyl moiety, namely a “push” mechanism, and a “pull” mechanism, based on the protonation of N7. Associated with the “push” mechanism and “pull” mechanisms are a series of proton transfer cascades, initiated from C8-H, via the tri-phosphate backbone, culminating in the formation of the pentavalent transition state between the g-phosphate of the ATP and the substrate nucleophile. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Bioprocess Optimization for the Production of Aromatic Compounds with Metabolically Engineered Hosts: Recent Developments and Future Challenges
fbioe-08-00096 February 18, 2020 Time: 17:51 # 1 REVIEW published: 20 February 2020 doi: 10.3389/fbioe.2020.00096 Bioprocess Optimization for the Production of Aromatic Compounds With Metabolically Engineered Hosts: Recent Developments and Future Challenges Adelaide Braga* and Nuno Faria Centre of Biological Engineering, University of Minho, Braga, Portugal The most common route to produce aromatic chemicals – organic compounds containing at least one benzene ring in their structure – is chemical synthesis. These processes, usually starting from an extracted fossil oil molecule such as benzene, Edited by: toluene, or xylene, are highly environmentally unfriendly due to the use of non- Nils Jonathan Helmuth Averesch, Stanford University, United States renewable raw materials, high energy consumption and the usual production of toxic Reviewed by: by-products. An alternative way to produce aromatic compounds is extraction from Nicolai Kallscheuer, plants. These extractions typically have a low yield and a high purification cost. Radboud University, Netherlands This motivates the search for alternative platforms to produce aromatic compounds Xiangzhao Mao, Ocean University of China, China through low-cost and environmentally friendly processes. Microorganisms are able to Mattijs K. Julsing, synthesize aromatic amino acids through the shikimate pathway. The construction of Wageningen University and Research, Netherlands microbial cell factories able to produce the desired molecule from renewable feedstock *Correspondence: becomes a promising alternative. This review article focuses on the recent advances Adelaide Braga in microbial production of aromatic products, with a special emphasis on metabolic [email protected] engineering strategies, as well as bioprocess optimization. The recent combination of Specialty section: these two techniques has resulted in the development of several alternative processes This article was submitted to to produce phenylpropanoids, aromatic alcohols, phenolic aldehydes, and others. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -

Prenylquinones in Human Parasitic Protozoa: Biosynthesis, Physiological Functions, and Potential As Chemotherapeutic Targets
molecules Review Prenylquinones in Human Parasitic Protozoa: Biosynthesis, Physiological Functions, and Potential as Chemotherapeutic Targets 1, 1, 1, 1,2 Ignasi B. Verdaguer y, Camila A. Zafra y, Marcell Crispim y, Rodrigo A.C. Sussmann , Emília A. Kimura 1 and Alejandro M. Katzin 1,* 1 Department of Parasitology, Institute of Biomedical Sciences, University of São Paulo, São Paulo 05508000, Brazil; [email protected] (I.B.V.); [email protected] (C.A.Z.); [email protected] (M.C.); [email protected] (R.A.C.S.); [email protected] (E.A.K.) 2 Centro de Formação em Ciências Ambientais, Universidade Federal do Sul da Bahia, Porto Seguro 45810-000 Bahia, Brazil * Correspondence: [email protected]; Tel.: +55-11-3091-7330; Fax: +5511-3091-7417 These authors contributed equally to this work. y Received: 5 August 2019; Accepted: 1 October 2019; Published: 16 October 2019 Abstract: Human parasitic protozoa cause a large number of diseases worldwide and, for some of these diseases, there are no effective treatments to date, and drug resistance has been observed. For these reasons, the discovery of new etiological treatments is necessary. In this sense, parasitic metabolic pathways that are absent in vertebrate hosts would be interesting research candidates for the identification of new drug targets. Most likely due to the protozoa variability, uncertain phylogenetic origin, endosymbiotic events, and evolutionary pressure for adaptation to adverse environments, a surprising variety of prenylquinones can be found within these organisms. These compounds are involved in essential metabolic reactions in organisms, for example, prevention of lipoperoxidation, participation in the mitochondrial respiratory chain or as enzymatic cofactors. -
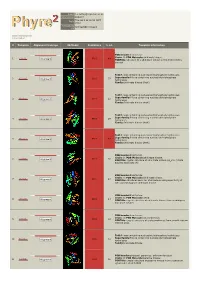
Phyre 2 Results for P0A6D7
Email [email protected] Description P0A6D7 Thu Jan 5 11:02:54 GMT Date 2012 Unique Job 0244e86604702aa1 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:transferase Chain: B: PDB Molecule:shikimate kinase; 1 c3trfB_ 99.9 44 Alignment PDBTitle: structure of a shikimate kinase (arok) from coxiella burnetii Fold:P-loop containing nucleoside triphosphate hydrolases Superfamily:P-loop containing nucleoside triphosphate 2 d2iyva1 99.9 37 Alignment hydrolases Family:Shikimate kinase (AroK) Fold:P-loop containing nucleoside triphosphate hydrolases Superfamily:P-loop containing nucleoside triphosphate 3 d1e6ca_ 99.9 32 Alignment hydrolases Family:Shikimate kinase (AroK) Fold:P-loop containing nucleoside triphosphate hydrolases Superfamily:P-loop containing nucleoside triphosphate 4 d1viaa_ 99.9 29 Alignment hydrolases Family:Shikimate kinase (AroK) Fold:P-loop containing nucleoside triphosphate hydrolases Superfamily:P-loop containing nucleoside triphosphate 5 d1kaga_ 99.9 97 Alignment hydrolases Family:Shikimate kinase (AroK) PDB header:transferase Chain: D: PDB Molecule:shikimate kinase; 6 c2pt5D_ 99.9 36 Alignment PDBTitle: crystal structure of shikimate kinase (aq_2177) from aquifex aeolicus2 vf5 PDB header:transferase Chain: A: PDB Molecule:shikimate kinase; 7 c1zuiA_ 99.9 29 Alignment PDBTitle: structural basis for shikimate-binding specificity of helicobacter2 pylori shikimate kinase PDB header:transferase Chain: B: PDB Molecule:atsk2; 8 c3nwjB_ 99.9 24 Alignment