Modeling Virus Transmission & Evolution in Mixed Communities
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mosquito Species Identification Using Convolutional Neural Networks With
www.nature.com/scientificreports OPEN Mosquito species identifcation using convolutional neural networks with a multitiered ensemble model for novel species detection Adam Goodwin1,2*, Sanket Padmanabhan1,2, Sanchit Hira2,3, Margaret Glancey1,2, Monet Slinowsky2, Rakhil Immidisetti2,3, Laura Scavo2, Jewell Brey2, Bala Murali Manoghar Sai Sudhakar1, Tristan Ford1,2, Collyn Heier2, Yvonne‑Marie Linton4,5,6, David B. Pecor4,5,6, Laura Caicedo‑Quiroga4,5,6 & Soumyadipta Acharya2* With over 3500 mosquito species described, accurate species identifcation of the few implicated in disease transmission is critical to mosquito borne disease mitigation. Yet this task is hindered by limited global taxonomic expertise and specimen damage consistent across common capture methods. Convolutional neural networks (CNNs) are promising with limited sets of species, but image database requirements restrict practical implementation. Using an image database of 2696 specimens from 67 mosquito species, we address the practical open‑set problem with a detection algorithm for novel species. Closed‑set classifcation of 16 known species achieved 97.04 ± 0.87% accuracy independently, and 89.07 ± 5.58% when cascaded with novelty detection. Closed‑set classifcation of 39 species produces a macro F1‑score of 86.07 ± 1.81%. This demonstrates an accurate, scalable, and practical computer vision solution to identify wild‑caught mosquitoes for implementation in biosurveillance and targeted vector control programs, without the need for extensive image database development for each new target region. Mosquitoes are one of the deadliest animals in the world, infecting between 250–500 million people every year with a wide range of fatal or debilitating diseases, including malaria, dengue, chikungunya, Zika and West Nile Virus1. -

Operations and Disease Surveillance
VECTOR CONTROL DISTRICT COUNTY OF SANTA CLARA MONTHLY REPORT MAY 2020 SAVE WATER. NOT MOSQUITOES. DON’T CREATE MOSQUITO HABITAT BY ACCIDENT. Over watering lawns and plants is a large contributor to mosquito breeding. Help fight the bite by: • Fixing leaky faucets and broken sprinklers. • Only apply as much waster as your plants and soil need. Visit SCCVector.org for more information. IN THIS ISSUE MANAGER'S MESSAGE 2 SERVICES AVAILABLE 3 OPERATIONS DATA 4 - 6 WEST NILE 7 - 8 SURVEILLANCE PUBLIC SURVICE 9 REQUESTS VECTOR CONTROL DISTRICT COUNTY OF SANTA CLARA CONSUMER & ENVIRONMENTAL PROTECTION AGENCY PAGE 2 | OPERATIONS AND SURVEILLANCE REPORT MESSAGE FROM THE MANAGER Warm weather is here and that means vectors such as ticks are more active. May was Lyme Disease Awareness Month, and the County of Santa Clara Vector Control District celebrated by participating and advertising the Centers for Disease Control and Prevention’s Lyme Disease Awareness Month Lunch and Learn Series. The weekly series offered interactive presentations on tick surveillance, tick collection, tick bite prevention, and other activities. The District reminds the public to practice preventative measures to avoid coming into contact with ticks. Use insect repellent approved by the Environmental Protection Agency, avoid areas with high grass, and check gear, pets, and children for ticks during and after being in tick habitat. Warm months also mean other vectors such as mosquitoes are more active as well. The District is hard at work monitoring for diseases, such as West Nile virus and Nayer Zahiri eliminating mosquito breeding in areas like marshes, neglected pools, curbs, catch basins, and ponds. -

The Mosquitoes of Alaska
LIBRAR Y ■JRD FEBE- Î961 THE U. s. DtPÁ¡<,,>^iMl OF AGidCÜLl-yí MOSQUITOES OF ALASKA Agriculture Handbook No. 182 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE U < The purpose of this handbook is to present information on the biology, distribu- tion, identification, and control of the species of mosquitoes known to occur in Alaska. Much of this information has been published in short papers in various journals and is not readily available to those who need a comprehensive treatise on this subject ; some of the material has not been published before. The information l)r()UKlit together here will serve as a guide for individuals and communities that have an interest and responsibility in mosquito problems in Alaska. In addition, the military services will have considerable use for this publication at their various installations in Alaska. CuUseta alaskaensis, one of the large "snow mosquitoes" that overwinter as adults and emerge from hiber- nation while much of the winter snow is on the ground. In some localities this species is suJBBciently abundant to cause serious annoy- ance. THE MOSQUITOES OF ALASKA By C. M. GJULLIN, R. I. SAILER, ALAN STONE, and B. V. TRAVIS Agriculture Handbook No. 182 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Washington, D.C. Issued January 1961 For «ale by the Superintendent of Document«. U.S. Government Printing Office Washington 25, D.C. - Price 45 cent» Contents Page Page History of mosquito abundance Biology—Continued and control 1 Oviposition 25 Mosquito literature 3 Hibernation 25 Economic losses 4 Surveys of the mosquito problem. 25 Mosquito-control organizations 5 Mosquito surveys 25 Life history 5 Engineering surveys 29 Eggs_". -

The Alameda County Mosquito Abatement District Control Program
THTHEE ALAMEDA CCOUNTYOUNTY MOSQMOSQUITO ABATEMENT DISTRICT CONTROL PROGRAM September 2011 Alameda County Mosquito Abatement District 23187 Connecticut Street Hayward, California 94545-1605 THE ALAMEDA COUNTY MOSQUITO ABATEMENT DISTRICT CONTROL PROGRAM Key Staff John Rusmisel - District Manager Bruce Kirkpatrick - Entomologist Erika Castillo - Environmental Specialist Patrick Turney - Field Supervisor Phone: 510-783-7744 Fax: 510-783-3903 email: [email protected] TABLE OF CONTENTS Mission and Vision Statement ......................................................................................................................................... 5 General Information about the District........................................................................................................................... 6 Location of the District ....................................................................................................................... 7 District Powers.................................................................................................................................... 7 The Value of an Organized Mosquito Control District ....................................................................... 8 District Goals and Services ................................................................................................................. 9 Service Calls ....................................................................................................................................... 10 District Finances ................................................................................................................................ -
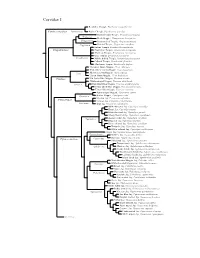
Corvidae Species Tree
Corvidae I Red-billed Chough, Pyrrhocorax pyrrhocorax Pyrrhocoracinae =Pyrrhocorax Alpine Chough, Pyrrhocorax graculus Ratchet-tailed Treepie, Temnurus temnurus Temnurus Black Magpie, Platysmurus leucopterus Platysmurus Racket-tailed Treepie, Crypsirina temia Crypsirina Hooded Treepie, Crypsirina cucullata Rufous Treepie, Dendrocitta vagabunda Crypsirininae ?Sumatran Treepie, Dendrocitta occipitalis ?Bornean Treepie, Dendrocitta cinerascens Gray Treepie, Dendrocitta formosae Dendrocitta ?White-bellied Treepie, Dendrocitta leucogastra Collared Treepie, Dendrocitta frontalis ?Andaman Treepie, Dendrocitta bayleii ?Common Green-Magpie, Cissa chinensis ?Indochinese Green-Magpie, Cissa hypoleuca Cissa ?Bornean Green-Magpie, Cissa jefferyi ?Javan Green-Magpie, Cissa thalassina Cissinae ?Sri Lanka Blue-Magpie, Urocissa ornata ?White-winged Magpie, Urocissa whiteheadi Urocissa Red-billed Blue-Magpie, Urocissa erythroryncha Yellow-billed Blue-Magpie, Urocissa flavirostris Taiwan Blue-Magpie, Urocissa caerulea Azure-winged Magpie, Cyanopica cyanus Cyanopica Iberian Magpie, Cyanopica cooki Siberian Jay, Perisoreus infaustus Perisoreinae Sichuan Jay, Perisoreus internigrans Perisoreus Gray Jay, Perisoreus canadensis White-throated Jay, Cyanolyca mirabilis Dwarf Jay, Cyanolyca nanus Black-throated Jay, Cyanolyca pumilo Silvery-throated Jay, Cyanolyca argentigula Cyanolyca Azure-hooded Jay, Cyanolyca cucullata Beautiful Jay, Cyanolyca pulchra Black-collared Jay, Cyanolyca armillata Turquoise Jay, Cyanolyca turcosa White-collared Jay, Cyanolyca viridicyanus -

Microsoft Outlook
Joey Steil From: Leslie Jordan <[email protected]> Sent: Tuesday, September 25, 2018 1:13 PM To: Angela Ruberto Subject: Potential Environmental Beneficial Users of Surface Water in Your GSA Attachments: Paso Basin - County of San Luis Obispo Groundwater Sustainabilit_detail.xls; Field_Descriptions.xlsx; Freshwater_Species_Data_Sources.xls; FW_Paper_PLOSONE.pdf; FW_Paper_PLOSONE_S1.pdf; FW_Paper_PLOSONE_S2.pdf; FW_Paper_PLOSONE_S3.pdf; FW_Paper_PLOSONE_S4.pdf CALIFORNIA WATER | GROUNDWATER To: GSAs We write to provide a starting point for addressing environmental beneficial users of surface water, as required under the Sustainable Groundwater Management Act (SGMA). SGMA seeks to achieve sustainability, which is defined as the absence of several undesirable results, including “depletions of interconnected surface water that have significant and unreasonable adverse impacts on beneficial users of surface water” (Water Code §10721). The Nature Conservancy (TNC) is a science-based, nonprofit organization with a mission to conserve the lands and waters on which all life depends. Like humans, plants and animals often rely on groundwater for survival, which is why TNC helped develop, and is now helping to implement, SGMA. Earlier this year, we launched the Groundwater Resource Hub, which is an online resource intended to help make it easier and cheaper to address environmental requirements under SGMA. As a first step in addressing when depletions might have an adverse impact, The Nature Conservancy recommends identifying the beneficial users of surface water, which include environmental users. This is a critical step, as it is impossible to define “significant and unreasonable adverse impacts” without knowing what is being impacted. To make this easy, we are providing this letter and the accompanying documents as the best available science on the freshwater species within the boundary of your groundwater sustainability agency (GSA). -

Operations and Disease Surveillance Report
Page 1 Santa Clara County Vector Control District Operations and Surveillance Report August 2018 District Mission Table of Contents page To detect and minimize vector-borne diseases, to abate mosquitoes, and to assist the public in resolving problems with rodents, wildlife, and insects that can cause disease, discomfort, or injury to humans in the County. Manager’s Message 1 Services Provided Operations Report: Curbs and Catch Basins 2 Detection of the presence/prevalence of vector-borne diseases, such as plague, West Nile virus, rabies, and Lyme disease, through ongoing surveillance and testing Routine inspections and treatment, as necessary, of Operations Report: known mosquito and rodent sources Neglected Pools 3 Response to customer initiated service requests for iden- and Mosquitofish tification, advice, and/or control measures for mosqui- toes, rodents, wildlife, and miscellaneous invertebrates (ticks, yellowjackets, cockroaches, bees, fleas, flies, etc.) Continuing Education at Free educational presentations for schools, homeowner Vector Control associations, private businesses, civic groups, and other 4 West Nile Virus interested organizations Surveillance Free informational material on all vectors and vector- borne diseases Carbon Dioxide Baited Traps 5 Manager’s Message West Nile Virus Update Mosquitofish is the common name for Gambusia affinis and is a small fish related to common guppies. They are called Public Service Requests mosquitofish because mosquito larvae are their primary diet Invasive Cockroach 6 and they can eat 100 to 500 mosquito larvae per day. Vector Detected in County Control District delivers free limited numbers of mosquitofish for residents’ pools or ponds. To request mosquitofish, please call or visit our website. Outreach Programs - 7 (408) 918-4770 West Nile Virus Treatments sccvector.org Page 2 Operations Report: Curbs and Catch Basins The District employs seasonal staff to check and treat mosquitoes in flooded street stormwater catch basins. -

Infection Dynamics and Immune Response in a Newly Described Mbio.Asm.Org Drosophila-Trypanosomatid Association on September 15, 2015 - Published by Phineas T
Downloaded from RESEARCH ARTICLE crossmark Infection Dynamics and Immune Response in a Newly Described mbio.asm.org Drosophila-Trypanosomatid Association on September 15, 2015 - Published by Phineas T. Hamilton,a Jan Votýpka,b,c Anna Dostálová,d Vyacheslav Yurchenko,c,e Nathan H. Bird,a Julius Lukeš,c,f,g Bruno Lemaitre,d Steve J. Perlmana,g Department of Biology, University of Victoria, Victoria, British Columbia, Canadaa; Department of Parasitology, Faculty of Sciences, Charles University, Prague, Czech Republicb; Biology Center, Institute of Parasitology, Czech Academy of Sciences, Budweis, Czech Republicc; Global Health Institute, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerlandd; Life Science Research Center, Faculty of Science, University of Ostrava, Ostrava, Czech Republice; Faculty of Science, University of South Bohemia, Budweis, Czech Republicf; Integrated Microbial Biodiversity Program, Canadian Institute for Advanced Research, Toronto, Ontario, Canadag ABSTRACT Trypanosomatid parasites are significant causes of human disease and are ubiquitous in insects. Despite the impor- tance of Drosophila melanogaster as a model of infection and immunity and a long awareness that trypanosomatid infection is common in the genus, no trypanosomatid parasites naturally infecting Drosophila have been characterized. Here, we establish a new model of trypanosomatid infection in Drosophila—Jaenimonas drosophilae, gen. et sp. nov. As far as we are aware, this is the first Drosophila-parasitic trypanosomatid to be cultured and characterized. Through experimental infections, we find that Drosophila falleni, the natural host, is highly susceptible to infection, leading to a substantial decrease in host fecundity. J. droso- mbio.asm.org philae has a broad host range, readily infecting a number of Drosophila species, including D. -

West Mexico: Tour Report 2017
WEST MEXICO: TOUR REPORT 2017 21st FEBRUARY – 9th MARCH TOUR HIGHLIGHTS: Either for rarity value, excellent views or simply a group favourite. • Dwarf Vireo • Rufous-bellied Chachalaca • GoldeN Vireo • Elegant Quail • San Blas Jay • LoNg-tailed Wood-Partridge • Tufted Jay • Rufous-necked Wood-Rail • Black-throated Magpie-Jay • Lesser RoadruNNer • Flammulated Flycatcher • Balsas Screech Owl • Grey Silky-Flycatcher • MexicaN Barred Owl • Spotted WreN • Colima Pygmy Owl • Blue Mockingbird • NortherN Potoo • BrowN-backed Solitaire • MexicaN Whip-poor-will • Russet NightiNgale-Thrush • Eared Poorwill • Olive Warbler • MexicaN Hermit • Crescent-chested Warbler • MexicaN Violetear • Colima Warbler • MexicaN WoodNymph • Red-faced Warbler • Bumblebee Hummingbird • GoldeN-browed Warbler • CitreoliNe TrogoN • Red Warbler • Coppery-tailed TrogoN • Collared Towhee • Russet-crowNed Motmot • Rusty-crowNed GrouNd Sparrow • GoldeN-cheeked Woodpecker • GreeN-striped Brush-FiNch • Lilac-crowNed AmazoN • Red-headed Tanager • MexicaN Parrotlet • Red-breasted Chat • Military Macaw • Varied BuNtiNg • ChestNut-sided Shrike-Vireo • OraNge-breasted Bunting • Black-capped Vireo SUMMARY: West Mexico is a birders paradise with a superb variety of habitats that harbour an excitiNg cast of endemics, along with an excellent supporting cast of amazing birds. Our tour produced 327 species seeN, of which 47 were MexicaN eNdemics. But it’s Not just about Numbers and the overall experieNce of seeing a wide variety of wiNteriNg warblers usually iN large flocks, some good shorebirds, aNd a wide variety of hummers, parrots and other really rare birds all in lovely warm sunshine certainly made this a very enjoyable tour. We travelled from Puerto Vallarta, where Blue MockiNgbird was a gardeN bird, aloNg the coast to VolcaN de Fuego aNd its sister, VolcaN de Nieve where we eNcouNtered Lesser RoadruNNer, Numerous OraNge-breasted Buntings and the fabulous Chestnut-sided Shrike-Vireo. -

The Entomopathogenic Fungusmetarhizium Anisopliae For
VWJS 1'J Ernst-Jan Scholte The entomopathogenic fungus Metarhiziumanisopliae for mosquito control Impact on the adult stage of the African malaria vector Anopheles gambiae and filariasis vector Culex quinquefasciatus Proefschrift terverkrijgin g vand egraa dva ndocto r opgeza gva n derecto r magnificus vanWageninge n Universiteit, Prof.Dr .Ir .L .Speelma n inhe topenbaa rt e verdedigen opdinsda g 30novembe r200 4 desnamiddag st ehal ftwe ei nd eAul a Scholte,Ernst-Ja n(2004 ) Theentomopathogeni cfungu sMetarhizium anisopliaefo r mosquitocontro l Impact onth e adult stage ofth eAfrica n malaria vectorAnopheles gambiae and filariasis vector Culex quinquefasciatus Thesis Wageningen University -withreferences - with summary inDutc h ISBN:90-8504-118- X fs/l0x£u Stellingen 1. Ondanks aanvankelijke scepsis kan deinsect-pathogen e schimmel Metarhizium anisopliaeworde n ingezet bij bestrijding van malaria muggen (dit proefschrift). 2. De ontdekking vanBacillus thuringiensis israelensis in 1976heef t het onderzoek naar, en ontwikkeling van, entomopathogeneschimmel s voorbestrijdin g van muggen sterk afgeremd (ditproefschrift) . 3. Biologische bestrijding van plagen isveilige r voor devolksgezondhei d dan chemische bestrijding (FrancisG . Howarthi nAnn. Rev. Entomol. 1991, 36: 485-509). 4. In etymologische zin kunnen muggen ook worden opgevat als amfibieen. 5. Het feit dat sommigeAfrikane n zich grappend afvragen of blanken welbene n hebben geeft aan dat de meesteblanke n zich tijdens eenbezoe k aan Afrika teweini g uithu n autobegeve n omzic honde r deplaatselijk e bevolking temengen . 6. Het isbiza r dat het land met het grootste wapenarsenaal, een land dat in de afgelopen 100jaa r bij 15oorloge n betrokken is geweest zonder dathe t land zelf ooitwer d binnengevallen, zich opwerpt alsbeschermenge l van dewereldvred e en democratie. -

Proposals 2020-C
AOS Classification Committee – North and Middle America Proposal Set 2020-C 2 March 2020 No. Page Title 01 02 Remove “Scrub” from the English names of the scrub-jays 02 07 Add Common Kingfisher Alcedo atthis to the Appendix, Part 1 03 09 Recognize four species as never established in Hawaii, resulting in (a) transfer of Red-cheeked Cordonbleu Uraeginthus bengalus from the main list to the Appendix, and (b) removal of Helmeted Guineafowl Numida meleagris, Black-rumped Waxbill Estrilda troglodytes, and Tricolored Munia Lonchura malacca from the list of species known to occur in the US 04 13 (a) Adopt the ABA-CLC criteria for considering species to be established, and (b) reconsider the status of four species currently accepted as established in the US: Japanese Quail Coturnix japonica, Mitred Parakeet Psittacara mitrata, Lavender Waxbill Estrilda caerulescens, and Orange-cheeked Waxbill E. melpoda 05 20 Revise species limits in the Zosterops japonicus complex 06 23 Change the genus of White-crowned Manakin from Dixiphia to Pseudopipra 07 26 Adopt West African Crested Tern as the English name for Thalasseus albididorsalis 08 27 Transfer Yellow-chevroned Parakeet Brotogeris chiriri from the Appendix to the main list 09 29 Change the species name of Dwarf Jay from Cyanolyca nana to C. nanus 10 30 Rectify the linear sequence of Progne spp. (Hirundinidae) 11 33 Transfer (a) Myrmeciza exsul to Poliocrania and M. laemosticta to Sipia, and (b) M. zeledoni to Hafferia 12 37 Revise the taxonomy of Paltry Tyrannulet Zimmerius vilissimus: (a) elevate extralimital subspecies improbus and petersi to species rank, (b) elevate subspecies parvus to species rank, and (c) adopt new English names in accordance with these changes 13 48 Split Dusky Thrush Turdus eunomus and Naumann’s Thrush T. -

Jewels of the Andes Rose Ann Rowlett
OCTOBER 2011 fieldguides® BIRDING TOURS WORLDWIDE Jewels of the Andes Rose Ann Rowlett Jewels indeed! One example from each of three important groups we seek on our Jewels of Ecuador tour: Giant Antpitta, Blue-winged Mountain-Tanager, and Violet-tailed Sylph. [Photos by guide Richard Webster & participant Ed Hagen] ike most North American birders, you’ve probably wandered toucan, probably Gray-breasted, for the dynamite colors on its bill! Then south from time to time, at least among the colorful plates of there’s the multicolored ToucanBarbet, with a bill so strange it has recently the many exciting books on the birds of South America. If you been accorded family rank (along with Prong-billed Barbet of Costa Rica and find the comfort of cooler temperatures appealing, you’ve Panama). You might throw in a dramatically beautiful woodpecker, maybe a probably been drawn to the birds of the Andean countries. If Powerful or a Crimson-mantled? How about a flock of Turquoise Jays, Lyou have yet to make the plunge, which species do you dream of seeing? gleaming blue as they hop along mossy branches? If you’re into raptors, you’ve What would you list as the classiest birds of the Andes? probably imagined a massive male Andean Condor circling in the sun below a Do you dream of Torrent Ducks repeatedly plunging from rocks into snow-capped volcanic peak…or a Black-and-chestnut Eagle diving for prey at rushing water and bounding back onto the rocks? Do you imagine a White- close range. If you love night birding, you may have imagined a spectacular cappedDipperhoppingthroughthemistatthebaseofatallwaterfall?Or male Lyre-tailed or Swallow-tailed Nightjar circling overhead, its tail streamers havetheshowierbirdsgrabbedyourattention?Howaboutatreefullof flowing in the spotlight.