COMPLETE INSTRUCTIONS for INSTALLING and RUNNING PC-GAMESS / FIREFLY UNDER MAC OS X (Tested on Tiger & Leopard) STEP 1 :: Ge
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ubuntu Kung Fu
Prepared exclusively for Alison Tyler Download at Boykma.Com What readers are saying about Ubuntu Kung Fu Ubuntu Kung Fu is excellent. The tips are fun and the hope of discov- ering hidden gems makes it a worthwhile task. John Southern Former editor of Linux Magazine I enjoyed Ubuntu Kung Fu and learned some new things. I would rec- ommend this book—nice tips and a lot of fun to be had. Carthik Sharma Creator of the Ubuntu Blog (http://ubuntu.wordpress.com) Wow! There are some great tips here! I have used Ubuntu since April 2005, starting with version 5.04. I found much in this book to inspire me and to teach me, and it answered lingering questions I didn’t know I had. The book is a good resource that I will gladly recommend to both newcomers and veteran users. Matthew Helmke Administrator, Ubuntu Forums Ubuntu Kung Fu is a fantastic compendium of useful, uncommon Ubuntu knowledge. Eric Hewitt Consultant, LiveLogic, LLC Prepared exclusively for Alison Tyler Download at Boykma.Com Ubuntu Kung Fu Tips, Tricks, Hints, and Hacks Keir Thomas The Pragmatic Bookshelf Raleigh, North Carolina Dallas, Texas Prepared exclusively for Alison Tyler Download at Boykma.Com Many of the designations used by manufacturers and sellers to distinguish their prod- ucts are claimed as trademarks. Where those designations appear in this book, and The Pragmatic Programmers, LLC was aware of a trademark claim, the designations have been printed in initial capital letters or in all capitals. The Pragmatic Starter Kit, The Pragmatic Programmer, Pragmatic Programming, Pragmatic Bookshelf and the linking g device are trademarks of The Pragmatic Programmers, LLC. -

Linux on the Road
Linux on the Road Linux with Laptops, Notebooks, PDAs, Mobile Phones and Other Portable Devices Werner Heuser <wehe[AT]tuxmobil.org> Linux Mobile Edition Edition Version 3.22 TuxMobil Berlin Copyright © 2000-2011 Werner Heuser 2011-12-12 Revision History Revision 3.22 2011-12-12 Revised by: wh The address of the opensuse-mobile mailing list has been added, a section power management for graphics cards has been added, a short description of Intel's LinuxPowerTop project has been added, all references to Suspend2 have been changed to TuxOnIce, links to OpenSync and Funambol syncronization packages have been added, some notes about SSDs have been added, many URLs have been checked and some minor improvements have been made. Revision 3.21 2005-11-14 Revised by: wh Some more typos have been fixed. Revision 3.20 2005-11-14 Revised by: wh Some typos have been fixed. Revision 3.19 2005-11-14 Revised by: wh A link to keytouch has been added, minor changes have been made. Revision 3.18 2005-10-10 Revised by: wh Some URLs have been updated, spelling has been corrected, minor changes have been made. Revision 3.17.1 2005-09-28 Revised by: sh A technical and a language review have been performed by Sebastian Henschel. Numerous bugs have been fixed and many URLs have been updated. Revision 3.17 2005-08-28 Revised by: wh Some more tools added to external monitor/projector section, link to Zaurus Development with Damn Small Linux added to cross-compile section, some additions about acoustic management for hard disks added, references to X.org added to X11 sections, link to laptop-mode-tools added, some URLs updated, spelling cleaned, minor changes. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Download Wineskin Wrapper
Download wineskin wrapper click here to download Wineskin Winery Wrapper Wine Xquartz X11 Mac OS X Macintosh free do-it-yourself porting microsoft windows programs run game games gaming.Downloads · Wineskin · Manual · Wineskin on OS X El Capitan. Manually Installed Item Downloads. If you'd like versions of Engines, Wrappers, etc you can install to Wineskin Winery manually instead of using its built in. If you want to actually install things to create wrappers for yourself or others. Wineskin wrappers are created in Wineskin Winery. Download the latest version of. Wineskin is a free, open source utility that ports Windows programs to OS X so you Step One: Download Wineskin and Update the Wrapper. Download Wineskin www.doorway.ru Version zip Features. Make Mac OS X ports/wrappers for Windows™ software; Integrated Wine. Wineskin Winery for Mac: Free Download - Make Wine wrappers to run Windows software on Mac OS X. Download the latest versions of the best Mac apps at. Simply download and extract the Wrapper –> download the rFactor Demo The engine isn't changed, only a new Icon and an update Wineskin version. Manage and download Engines, or even custom build engines from Wine source code. Get Master Wrapper updates Create Wineskin wrappers Wineskin is a. Once the the installers finish downloading, it will create your wrapper. By default, it is stored in Applications > Wineskin > [your wrapper]. Then it. Before you even make the wrapper you'll need to pick a game you want to port to the goto this website to download wineskin Winery for Mac. To sum up: Download the game. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -

Bringing Windows to the Linux Desktop
BRINGING WINDOWS TO THE LINUX DESKTOP In a perfect world, the operating system that you use wouldn't matter. We're moving towards that with the growing use and availability of Web applications. But the day when the operating system is irrelevant is still awhile off. Right now, though, Windows is the dominant operating system. And there are no Linux versions of the popular and most widely-used applications for Windows -- programs like Microsoft Office and Adobe Photoshop. If you're a Linux user and you need to play nicely with the Windows world, conventional wisdom dictates that you either need a separate desktop computer or laptop computer that's running Windows or a system that dual boots. If you're adventurous, you can install a virtualization tool like VM Ware or Virtual Box. This software recreates Windows on your Linux box. The only problem with those solutions is that they need a lot of hard drive space and CPU. There is another choice: fake it. Faking it What do I mean by faking it? Using software that fools Windows applications into believing that they're actually running on Windows and not on Linux. It does this by creating what's called anapplication layer between the Windows program and Linux. The application layer simulates the Windows environment -- calls to the operating system, DLLs, drivers, and the like. Unlike the other solutions mentioned earlier, using an application layer only adds a bit of extra load to your computer -- a few megabytes of disk space and a couple of megabytes of RAM. -
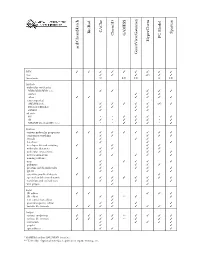
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Linux Networking Cookbook.Pdf
Linux Networking Cookbook ™ Carla Schroder Beijing • Cambridge • Farnham • Köln • Paris • Sebastopol • Taipei • Tokyo Linux Networking Cookbook™ by Carla Schroder Copyright © 2008 O’Reilly Media, Inc. All rights reserved. Printed in the United States of America. Published by O’Reilly Media, Inc., 1005 Gravenstein Highway North, Sebastopol, CA 95472. O’Reilly books may be purchased for educational, business, or sales promotional use. Online editions are also available for most titles (safari.oreilly.com). For more information, contact our corporate/institutional sales department: (800) 998-9938 or [email protected]. Editor: Mike Loukides Indexer: John Bickelhaupt Production Editor: Sumita Mukherji Cover Designer: Karen Montgomery Copyeditor: Derek Di Matteo Interior Designer: David Futato Proofreader: Sumita Mukherji Illustrator: Jessamyn Read Printing History: November 2007: First Edition. Nutshell Handbook, the Nutshell Handbook logo, and the O’Reilly logo are registered trademarks of O’Reilly Media, Inc. The Cookbook series designations, Linux Networking Cookbook, the image of a female blacksmith, and related trade dress are trademarks of O’Reilly Media, Inc. Java™ is a trademark of Sun Microsystems, Inc. .NET is a registered trademark of Microsoft Corporation. Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. Where those designations appear in this book, and O’Reilly Media, Inc. was aware of a trademark claim, the designations have been printed in caps or initial caps. While every precaution has been taken in the preparation of this book, the publisher and author assume no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. -

Cryptographic File Systems Performance: What You Don't Know Can Hurt You Charles P
Cryptographic File Systems Performance: What You Don't Know Can Hurt You Charles P. Wright, Jay Dave, and Erez Zadok Stony Brook University Appears in the proceedings of the 2003 IEEE Security In Storage Workshop (SISW 2003) Abstract interact with disks, caches, and a variety of other com- plex system components — all having a dramatic effect Securing data is more important than ever, yet cryp- on performance. tographic file systems still have not received wide use. In this paper we perform a real world performance One barrier to the adoption of cryptographic file systems comparison between several systems that are used is that the performance impact is assumed to be too high, to secure file systems on laptops, workstations, and but in fact is largely unknown. In this paper we first moderately-sized file servers. We also emphasize multi- survey available cryptographic file systems. Second, programming workloads, which are not often inves- we perform a performance comparison of a representa- tigated. Multi-programmed workloads are becoming tive set of the systems, emphasizing multiprogrammed more important even for single user machines, in which workloads. Third, we discuss interesting and counterin- Windowing systems are often used to run multiple appli- tuitive results. We show the overhead of cryptographic cations concurrently. We expect cryptographic file sys- file systems can be minimal for many real-world work- tems to become a commodity component of future oper- loads, and suggest potential improvements to existing ating systems. systems. We have observed not only general trends with We present results from a variety of benchmarks, an- each of the cryptographic file systems we compared but alyzing the behavior of file systems for metadata op- also anomalies based on complex interactions with the erations, raw I/O operations, and combined with CPU operating system, disks, CPUs, and ciphers.