Universal and Domain-Specific Sequences in 23S–28S Ribosomal RNA Identified by Computational Phylogenetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Final Copy 2018 09 25 Gaunt
This electronic thesis or dissertation has been downloaded from Explore Bristol Research, http://research-information.bristol.ac.uk Author: Gaunt, Jess Title: A Viral Approach to Translatome Profiling of CA1 Neurons During Associative Recognition Memory Formation General rights Access to the thesis is subject to the Creative Commons Attribution - NonCommercial-No Derivatives 4.0 International Public License. A copy of this may be found at https://creativecommons.org/licenses/by-nc-nd/4.0/legalcode This license sets out your rights and the restrictions that apply to your access to the thesis so it is important you read this before proceeding. Take down policy Some pages of this thesis may have been removed for copyright restrictions prior to having it been deposited in Explore Bristol Research. However, if you have discovered material within the thesis that you consider to be unlawful e.g. breaches of copyright (either yours or that of a third party) or any other law, including but not limited to those relating to patent, trademark, confidentiality, data protection, obscenity, defamation, libel, then please contact [email protected] and include the following information in your message: •Your contact details •Bibliographic details for the item, including a URL •An outline nature of the complaint Your claim will be investigated and, where appropriate, the item in question will be removed from public view as soon as possible. A Viral Approach to Translatome Profiling of CA1 Neurons During Associative Recognition Memory Formation Jessica Ruth Gaunt A dissertation submitted to the University of Bristol in accordance with the requirements for award of the degree of Doctor of Philosophy in the Faculty of Health Sciences, Bristol Medical School. -

Supplementary Figure 1
Supplementary Table 1 siRNA Oligonucleotide Sequences not Used for IGFBP-3 Knockdown siRNA Sequence nucleotides Source GCUACAAAGUUGACUACGA 686-704 ON-TARGET Plus SMART pool sequences GAAAUGCUAGUGAGUCGGA 536-554 ON-TARGET Plus SMART pool sequences GCACAGAUACCCAGAACUU 713-731 ON-TARGET Plus SMART pool sequences GAAUAUGGUCCCUGCCGUA 757-775 ON-TARGET Plus SMART pool sequences UAUCGAGAAUAGGAAAACC 1427-1445 siDESIGN center GCAGCCUCUCCCAGGCUACA 940-958 siDESIGN center GCAUAAGCUCUUUAAAGGCA 1895-1913 siDESIGN center UGCCUGGAUUCCACAGCUU 44-62 siDESIGN center AAGCAGCGTGCCCCGGUUG 106-124 siDESIGN center AAAGGCAAAGCUUUAUUUU 1908-1926 siDESIGN center Oligonucleotide sequences used for siRNA oligonucleotides tested to induce IGFBP-3 knockdown. Sequences 1-4 were from ON-TARGET Plus SMART pool sequences (Cat. # L-004777-00-0005, Dharmacon, Lafayette, CO). Sequences 5-10 were generated in our laboratory using the siDESIGN center from the Dharmacon website (www.dharmacon.com) by inputting the Genbank accession number NM_000598 (IGFBP-3). Supplementary Table 2 Transcripts Activated by NKX3.1 in PC-3 Cells PC-3 cells were stably transfected with the pcDNA3.1 empty vector or NKX3.1 expression vector and mRNA from two clones of each cell type was isolated for microarray analysis on the Affymetrix U-133 expression array. Analyses of results from each pair of clones of the same genotype that did not match up were discarded to ensure clonal variation was not a factor. 984 genes were found to be up- or down-regulated more that 1.4 fold in the NKX3.1 expressing PC-3 cells, in comparison to the PC-3 control cells. The 6th and 9th most activated probe sets were for human growth hormone-dependent insulin-like growth factor-binding protein, now known as IGFBP-3. -

Constructing and Analyzing Biological Interaction Networks for Knowledge Discovery
Constructing and Analyzing Biological Interaction Networks for Knowledge Discovery Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Duygu Ucar Graduate Program in Computer Science and Engineering The Ohio State University 2009 Dissertation Committee: Srinivasan Parthasarathy, Advisor Yusu Wang Umit Catalyurek c Copyright by Duygu Ucar 2009 ABSTRACT Many biological datasets can be effectively modeled as interaction networks where nodes represent biological entities of interest such as proteins, genes, or complexes and edges mimic associations among them. The study of these biological network structures can provide insight into many biological questions including the functional characterization of genes and gene products, the characterization of DNA-protein bindings, and the under- standing of regulatory mechanisms. Therefore, the task of constructing biological interac- tion networks from raw data sets and exploiting information from these networks is critical, but is also fraught with challenges. First, the network structure is not always known in a priori; the structure should be inferred from raw and heterogeneous biological data sources. Second, biological networks are noisy (containing unreliable interactions) and incomplete (missing real interactions) which makes the task of extracting useful information difficult. Third, typically these networks have non-trivial topological properties (e.g., uneven degree distribution, small world) that limit the effectiveness of traditional knowledge discovery al- gorithms. Fourth, these networks are usually dynamic and investigation of their dynamics is essential to understand the underlying biological system. In this thesis, we address these issues by presenting a set of computational techniques that we developed to construct and analyze three specific types of biological interaction networks: protein-protein interaction networks, gene co-expression networks, and regulatory networks. -

Multifaceted Deregulation of Gene Expression and Protein Synthesis with Age
Multifaceted deregulation of gene expression and protein synthesis with age Aleksandra S. Anisimovaa,b,c,1, Mark B. Meersona,c, Maxim V. Gerashchenkob, Ivan V. Kulakovskiya,d,e,f,2, Sergey E. Dmitrieva,c,d,2, and Vadim N. Gladyshevb,2 aBelozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; bDivision of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; cFaculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow 119234, Russia; dEngelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow 119991, Russia; eVavilov Institute of General Genetics, Russian Academy of Sciences, Moscow 119991, Russia; and fInstitute of Protein Research, Russian Academy of Sciences, Pushchino 142290, Russia Edited by Joseph D. Puglisi, Stanford University School of Medicine, Stanford, CA, and approved May 27, 2020 (received for review January 30, 2020) Protein synthesis represents a major metabolic activity of the cell. RNA polymerase I, eIF2Be, and eEF2, decrease with age in rat However, how it is affected by aging and how this in turn impacts tissues (10). Increased promoter methylation in ribosomal RNA cell function remains largely unexplored. To address this question, genes and decreased ribosomal RNA concentration during aging herein we characterized age-related changes in both the transcrip- were also reported (11). In addition, down-regulation of trans- tome and translatome of mouse tissues over the entire life span. lation with age was confirmed in vivo in the sheep (12) as well as We showed that the transcriptome changes govern those in the in replicatively aged yeast (13). -

(12) United States Patent (10) Patent No.: US 9,476,099 B2 Spinella Et Al
US009476.099B2 (12) United States Patent (10) Patent No.: US 9,476,099 B2 Spinella et al. (45) Date of Patent: Oct. 25, 2016 (54) METHOD FOR DETERMINING FOREIGN PATENT DOCUMENTS SENSTIVITY TO DECTABINE WO WO 2012/031008 A2 3, 2012 TREATMENT WO WO 2012, 11885.6 A1 9, 2012 (71) Applicant: TRUSTEES OF DARTMOUTH COLLEGE, Hanover, NH (US) OTHER PUBLICATIONS (72) Inventors: Michael Spinella, Hanover, NH (US); Tsai et al Cancer Cell. Mar. 20, 2012. 21(3):43.0-446 and Supple Maroun J. Beyrouthy, Lebanon, NH mental pages 1-18. (US) Agilent Technologies. DNA Oligo Microarray Gene Lists and Annotations, available via url: <chem.agilent.com/cag?bSp? gene (73) Assignee: Trustees of Dartmouth College, lists.asp printed on Mar. 1, 2016.* Hanover, NH (US) Abele et al. “The EORTC Early Clinical Trials Cooperative Group Experience with 5-Aza-2'-deoxycytidine (NSC 127716) in Patients (*) Notice: Subject to any disclaimer, the term of this with Colo-rectal, Head and Neck, Renal Carcinomas and Malignant patent is extended or adjusted under 35 Melanomas' European Journal of Cancer and Clinical Oncology U.S.C. 154(b) by 0 days. 1987 23(12): 1921-1924. Adewumi et al. “Characterization of Human Embryonic Stem Cell (21) Appl. No.: 14/416,142 Lines by the International StemCell Initiative” Nature Biotechnol ogy 2007 vol. 25(7):803-816. (22) PCT Filed: Jul. 31, 2013 Al-Hajj et al. "Prospective Identification of Tumorigenic Breast Cancer Cells' Proceedings of the National Academy of Sciences (86). PCT No.: PCT/US2013/052899 2003 100(7):3983-3988 with correction. -

Dissertation
Dissertation submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the degree of Doctor of Natural Sciences Presented by Dipl. Biochemist Birgit Rogell born in Speyer (Germany) nd Oral-examination: 2 November 2016 Exploring the biology of RNPs: specific capture of RNPs using antisense locked nucleic acids Referees: 1. Dr. Anne Ephrussi 2. Prof. Dr. Ralf Bartenschlager Abstract Abstract RNA-binding proteins (RBPs) are central players in cell biology and respond to a multitude of cellular cues and environmental stimuli. Identification of RBPs associated with specific transcripts in a cell is a challenging task; and the available strategies to purify specific transcripts and their bound proteome face numerous limitations. Thus, methods to determine the composition of proteins on a given RNA are required to further understand the regulation and biological function of any given RNA. Therefore, the focus of my PhD project was to develop a highly specific and selective method, “specific Ribonucleoprotein (RNP) capture”, to isolate a specific RNA species together with its bound proteome. Following irradiation with ultraviolet (UV) light that creates a covalent bond between RNA and protein, RNAs are captured using short LNA (locked nucleic acid)/DNA mixmer antisense probes coupled to a solid support.The proteins covalently linked to the isolated RNA are then identified by quantitative mass spectrometry. First, I successfully established the method for this application in vitro. Mass spectrometry data revealed that the protein Sister of Sex lethal (Ssx) has similar binding preferences to a mRNA derived from male-specific lethal (msl)2 mRNA as its paralog Sxl in Drosophila melanogaster embryo extracts. -

Expression of Distinct Maternal and Somatic 5.8S, 18S, and 28S Rrna Types During Zebrafish Development
Downloaded from rnajournal.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press REPORT Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development MAURO D. LOCATI,1,3 JOHANNA F.B. PAGANO,1,3 GENEVIÈVE GIRARD,1 WIM A. ENSINK,1 MARINA VAN OLST,1 SELINA VAN LEEUWEN,1 ULRIKE NEHRDICH,2 HERMAN P. SPAINK,2 HAN RAUWERDA,1 MARTIJS J. JONKER,1 ROB J. DEKKER,1 and TIMO M. BREIT1 1RNA Biology and Applied Bioinformatics Research Group, Swammerdam Institute for Life Sciences, Faculty of Science, University of Amsterdam, Amsterdam 1090 GE, the Netherlands 2Department of Molecular Cell Biology, Institute of Biology, Leiden University, Gorlaeus Laboratories–Cell Observatorium, Leiden 2333 CE, the Netherlands ABSTRACT There is mounting evidence that the ribosome is not a static translation machinery, but a cell-specific, adaptive system. Ribosomal variations have mostly been studied at the protein level, even though the essential transcriptional functions are primarily performed by rRNAs. At the RNA level, oocyte-specific 5S rRNAs are long known for Xenopus. Recently, we described for zebrafish a similar system in which the sole maternal-type 5S rRNA present in eggs is replaced completely during embryonic development by a somatic-type. Here, we report the discovery of an analogous system for the 45S rDNA elements: 5.8S, 18S, and 28S. The maternal-type 5.8S, 18S, and 28S rRNA sequences differ substantially from those of the somatic-type, plus the maternal-type rRNAs are also replaced by the somatic-type rRNAs during embryogenesis. We discuss the structural and functional implications of the observed sequence differences with respect to the translational functions of the 5.8S, 18S, and 28S rRNA elements. -

Meta-Type Analysis of Dopaminergic Effects on Gene Expression in the Neuroendocrine Brain of Female Goldfish
ORIGINAL RESEARCH ARTICLE published: 02 November 2012 doi: 10.3389/fendo.2012.00130 Meta-type analysis of dopaminergic effects on gene expression in the neuroendocrine brain of female goldfish JasonT. Popesku 1*†, Christopher J. Martyniuk 2 and Vance L.Trudeau 1* 1 Centre for Advanced Research in Environmental Genomics, Department of Biology, University of Ottawa, Ottawa, ON, Canada 2 Canadian Rivers Institute and Department of Biology, University of New Brunswick, Saint John, NB, Canada Edited by: Dopamine (DA) is a major neurotransmitter important for neuroendocrine control and recent Wei Ge, The Chinese University of studies have described genomic signaling pathways activated and inhibited by DA agonists Hong Kong, China and antagonists in the goldfish brain. Here we perform a meta-type analysis using microar- Reviewed by: José A. Muñoz-Cueto, University of ray datasets from experiments conducted with female goldfish to characterize the gene Cadiz, Spain expression responses that underlie dopaminergic signaling. Sexually mature, pre-spawning Anderson O. Wong, The University of [gonadosomatic index (GSI) D 4.5 ± 1.3%] or sexually regressing (GSI D 3 ± 0.4%) female Hong Kong, Hong Kong goldfish (15–40 g) injected intraperitoneally with either SKF 38393, LY 171555, SCH 23390, *Correspondence: sulpiride, or a combination of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and a-methyl-p- Jason T. Popesku, Centre for Advanced Research in Environmental tyrosine. Microarray meta-type analysis identified 268 genes in the telencephalon and hypo- Genomics, Department of Biology, thalamus as having reciprocal (i.e., opposite between agonism and antagonism/depletion) University of Ottawa, Ottawa, ON, fold change responses, suggesting that these transcripts are likely targets for DA-mediated Canada K1N 6N5. -

Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM
molecules Review Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM Frédéric Poitevin 1 , Artem Kushner 2,3, Xinpei Li 2,3 and Khanh Dao Duc 2,3,4,* 1 Department of LCLS Data Analytics, Linac Coherent Light Source, SLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA; [email protected] 2 Department of Mathematics, University of British Columbia, Vancouver, BC V6T 1Z4, Canada; [email protected] (A.K.); [email protected] (X.L.) 3 Department of Computer Science, University of British Columbia, Vancouver, BC V6T 1Z4, Canada 4 Department of Zoology, University of British Columbia, Vancouver, BC V6T 1Z4, Canada * Correspondence: [email protected] Academic Editor: Quentin Vicens Received: 25 August 2020; Accepted: 15 September 2020; Published: 17 September 2020 Abstract: The extent of ribosomal heterogeneity has caught increasing interest over the past few years, as recent studies have highlighted the presence of structural variations of the ribosome. More precisely, the heterogeneity of the ribosome covers multiple scales, including the dynamical aspects of ribosomal motion at the single particle level, specialization at the cellular and subcellular scale, or evolutionary differences across species. Upon solving the ribosome atomic structure at medium to high resolution, cryogenic electron microscopy (cryo-EM) has enabled investigating all these forms of heterogeneity. In this review, we present some recent advances in quantifying ribosome heterogeneity, with a focus on the conformational and evolutionary variations of the ribosome and their functional implications. These efforts highlight the need for new computational methods and comparative tools, to comprehensively model the continuous conformational transition pathways of the ribosome, as well as its evolution. -

Jlhulm.Pdf (2.103Mb)
CHARACTERIZATION OF THE FOUR GENES ENCODING CYTOPLASMIC RIBOSOMAL PROTEIN S15a IN ARABIDOPSIS THALIANA A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Biology University of Saskatchewan Saskatoon By Jacqueline Louise Hulm © Copyright Jacqueline Louise Hulm, March, 2008. All rights reserved. PERMISSION TO USE In presenting this thesis in partial fulfillment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by the professor or professors who supervised my thesis work or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or in part should be addressed to: Head of the Department of Biology 112 Science Place, University of Saskatchewan Saskatoon, Saskatchewan S7N 5E2 i ABSTRACT Eukaryotic cytosolic ribosomes are composed of two distinct subunits consisting of four individual ribosomal RNAs and, in Arabidopsis thaliana, 81 ribosomal proteins. Functional subunit assembly is dependent on the equimolar production of each ribosomal component. -
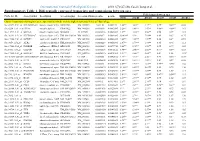
Differentially Expressed Transcripts and Comparisons Between Ages
International Journal of Biological Sciences 2010; 6(7):627-638, Caode Jiang et al. Supplementary Table 1. Differentially expressed transcripts and comparisons between ages Fold changes between ages Probe Set ID Gene Symbol Description Accession (pig) Accession (Human)p-value q-value d7/d0 d14/d0 d21/d0 d14/d7 d21/d7 d21/d14 Cluster 1:transcripts showing increased expression after birth, with the highest expression levels at 7 days of age Ssc.24673.1.S1_at HIST2H2AA4 histone cluster 2 H2aa4BX923965 NM_003516 1.22E-06 0.004472 4.88** 3.85** 3.17** 0.79* 0.65** 0.82 Ssc.6258.1.A1_a_at ITGA6 integrin alpha 6 CK463022 NM_000210 0.0000111 0.0063885 2.26** 1.49** 1.53** 0.66** 0.68** 1.03 Ssc.11712.1.A1_at KIF26A kinesin family memberBI184001 26A AL117589 0.0000126 0.0063885 3.1** 3.03** 3.06** 0.98 0.99 1.01 Ssc.26292.1.S1_at HIST1H2AC histone cluster 1 H2acXM_001928124 NM_003512 0.000017 0.0063885 10.42** 9.3* 7.01** 0.89 0.67 0.75 Ssc.17186.2.S1_at IGFBP7 insulin-like growth factorCR626227 binding proteinNM_001553 7 0.0000183 0.0063885 2.48** 2.09** 1.99** 0.84 0.8* 0.95 Ssc.3059.1.S1_at ALR2 aldo-keto reductase familyNM_001001539 1 member B1NM_003857 0.0000219 0.0066896 2.07** 1.79** 1.98** 0.86 0.96 1.11 Ssc.17186.2.S2_at POLR2B polymerase (RNA) II AF265351(DNA directed) polypeptideNM_000938 B,140kDa0.0000319 0.0077954 2.02** 1.92** 1.96** 0.95 0.97 1.02 Ssc.6435.1.S1_at COL4A1 collagen type IV alphaCN154120 1 NM_001845 0.0000339 0.0078122 3.44** 2.46** 2.05** 0.71* 0.6** 0.83 Ssc.17942.1.A1_at ELOVL7 ELOVL family memberCF176409 7 elongation -

Tetrapod Phylogeny Inferred from 18S and 28S Ribosomal RNA Sequences and a Review of the Evidence for Amniote Relationships1 S
Tetrapod Phylogeny Inferred from 18s and 28s Ribosomal RNA Sequences and a Review of the Evidence for Amniote Relationships1 S. Blair Hedgest * Kirk D. Moberg,? and Linda R. Maxson* *Department of Biology and Institute of Molecular Evolutionary Genetics, Pennsylvania State University, and ?University of Illinois College of Medicine at Urbana-Champaign The 18s ribosomal RNAs of 21 tetrapods were sequenced and aligned with five published tetrapod sequences. When the coelacanth was used as an outgroup, Lis- samphibia (living amphibians) and Amniota (amniotes) were found to be statis- tically significant monophyletic groups. Although little resolution was obtained among the lissamphibian taxa, the amniote sequences support a sister-group rela- tionship between birds and mammals. Portions of the 28s ribosomal RNA (rRNA) molecule in 11 tetrapods also were sequenced, although the phylogenetic results were inconclusive. In contrast to previous studies, deletion or down-weighting of base-paired sites were found to have little effect on phylogenetic relationships. Mo- lecular evidence for amniote relationships is reviewed, showing that three genes (beta-hemoglobin, myoglobin, and 18s rRNA) unambiguously support a bird- mammal relationship, compared with one gene (histone H2B) that favors a bird- crocodilian clade. Separate analyses of four other genes (alphacrystallin A, alpha- hemoglobin, insulin, and 28s rRNA) and a combined analysis of all sequence data are inconclusive, in that different groups are defined in different analyses and none are strongly supported. It is suggested that until sequences become available from a broader array of taxa, the molecular evidence is best evaluated at the level of individual genes, with emphasis placed on those studies with the greatest number of taxa and sites.