NIH Public Access Author Manuscript Nat Rev Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
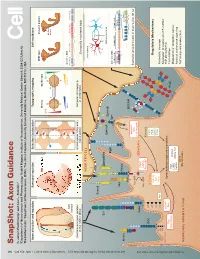
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Characterization of Increasing Stages of Invasiveness Identifies Stromal/Cancer Cell Crosstalk in Rat Models of Mesothelioma
www.oncotarget.com Oncotarget, 2018, Vol. 9, (No. 23), pp: 16311-16329 Research Paper Characterization of increasing stages of invasiveness identifies stromal/cancer cell crosstalk in rat models of mesothelioma Joëlle S. Nader1, Jérôme Abadie1,2, Sophie Deshayes1, Alice Boissard3,4, Stéphanie Blandin5, Christophe Blanquart1, Nicolas Boisgerault1, Olivier Coqueret3,4, Catherine Guette3,4, Marc Grégoire1 and Daniel L. Pouliquen1 1CRCINA, INSERM, Université d’Angers, Université de Nantes, Nantes, France 2ONIRIS, Nantes, France 3CRCINA, INSERM, Université de Nantes, Université d’Angers, Angers, France 4ICO, Angers, France 5Plate-Forme MicroPICell, SFR François Bonamy, Université de Nantes, France Correspondence to: Daniel L. Pouliquen, email: [email protected] Keywords: stroma; invasiveness; sarcomatoid mesothelioma; immune cells; rat model Received: November 08, 2017 Accepted: February 25, 2018 Published: March 27, 2018 Copyright: Nader et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Sarcomatoid mesothelioma (SM) is a devastating cancer associated with one of the poorest outcome. Therefore, representative preclinical models reproducing different tumor microenvironments (TME) observed in patients would open up new prospects for the identification of markers and evaluation of innovative therapies. Histological analyses -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Mediated Enamel Mineralization
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archivio della ricerca- Università di Roma La Sapienza [Frontiers In Bioscience, Landmark, 22, 1289-1329, March 1, 2017] The crossroads between cancer immunity and autoimmunity: antibodies to self antigens Monica Benvenuto1, Rosanna Mattera1, Laura Masuelli2, Ilaria Tresoldi1, Maria Gabriella Giganti1, Giovanni Vanni Frajese3, Vittorio Manzari1, Andrea Modesti1,4, Roberto Bei1,4 1Department of Clinical Sciences and Translational Medicine, University of Rome, Tor Vergata, Rome, 00133 Italy, 2Department of Experimental Medicine, University of Rome, Sapienza, Rome, 00164 Italy, 3Department of Sports Science, Human and Health, University of Rome ‘Foro Italico’, Rome, 00135 Italy, 4Center for Regenerative Medicine, (CIMER), University of Rome “Tor Vergata”, Rome, 00133 Italy TABLE OF CONTENTS 1. Abstract 2. Introduction 3. Immune response to self antigens in cancer patients partly covers that characteristic of patients with autoimmune diseases 4. Features of self antigens involved in the autoreactive immune response: immunological properties and abnormal expression 5. Biological activities of autoantibodies to self antigens 6. Association of autoantibodies to self antigens with paraneoplastic autoimmune syndromes 7. Role of autoantibodies to self antigens as biomarkers for cancer detection and cancer patients prognosis 8. Paraneoplastic neurological syndromes, effects of therapy targeting immune-checkpoint receptors and Tregs dysregulation in autoimmune disease patients: the crossroad between autoimmunity and immune response in cancer patients 9. Conclusions 10. Acknowledgment 11. References 1. ABSTRACT The production of autoantibodies to self meaning of autoantibodies to self antigens detected in antigens is dependent on the failure of immune cancer and autoimmune disease patients. -

Vaccines and Other Immunological Approaches for Cancer Immunoprevention
Current Drug Targets, 2011, 12, 1957-1973 1957 Vaccines and Other Immunological Approaches for Cancer Immunoprevention Pier-Luigi Lollini*,1, Giordano Nicoletti2, Lorena Landuzzi2, Federica Cavallo3, Guido Forni3, Carla De Giovanni4 and Patrizia Nanni4 1Department of Hematology and Oncological Sciences, University of Bologna; 2Rizzoli Orthopedic Institute, Bologna; 3Molecular Biotechnology Center, Department of Clinical and Biological Sciences, University of Turin; 4Department of Experimental Pathology, University of Bologna, Italy Abstract: The immune system effectively prevents cancer, whereas severe immunodepression increases its incidence. Cancer immunoprevention is a strategy based on the concept that enhancement of tumor immunity in healthy individuals reduces cancer risk. It can be viewed as a kind of chemoprevention. For cancer immunoprevention, the cancer universe can be neatly divided between tumors caused - directly or indirectly - by infectious agents and all other tumors. Immunoprevention of tumors caused by infectious agents is already implemented at the population level for hepatitis B virus (HBV)-related hepatocellular carcinoma and for tumors caused by human papillomaviruses (HPV), like cervical carcinoma. Now the challenge is to develop immunological strategies to prevent the bulk (>80%) of human tumor burden, unrelated to infections. Both vaccines against tumor antigens and immune modulators can prevent tumor onset in cancer- prone mice. These studies outlined the target antigens and the molecular and cellular mechanisms -

Supplemental Tables4.Pdf
Yano_Supplemental_Table_S4 Gene ontology – Biological process 1 of 9 Fold List Pop Pop GO Term Count % PValue Bonferroni Benjamini FDR Genes Total Hits Total Enrichment DLC1, CADM1, NELL2, CLSTN1, PCDHGA8, CTNNB1, NRCAM, APP, CNTNAP2, FERT2, RAPGEF1, PTPRM, MPDZ, SDK1, PCDH9, PTPRS, VEZT, NRXN1, MYH9, GO:0007155~cell CTNNA2, NCAM1, NCAM2, DDR1, LSAMP, CNTN1, 50 5.61 2.14E-08 510 311 7436 2.34 4.50E-05 4.50E-05 3.70E-05 adhesion ROR2, VCAN, DST, LIMS1, TNC, ASTN1, CTNND2, CTNND1, CDH2, NEO1, CDH4, CD24A, FAT3, PVRL3, TRO, TTYH1, MLLT4, LPP, NLGN1, PCDH19, LAMA1, ITGA9, CDH13, CDON, PSPC1 DLC1, CADM1, NELL2, CLSTN1, PCDHGA8, CTNNB1, NRCAM, APP, CNTNAP2, FERT2, RAPGEF1, PTPRM, MPDZ, SDK1, PCDH9, PTPRS, VEZT, NRXN1, MYH9, GO:0022610~biological CTNNA2, NCAM1, NCAM2, DDR1, LSAMP, CNTN1, 50 5.61 2.14E-08 510 311 7436 2.34 4.50E-05 4.50E-05 3.70E-05 adhesion ROR2, VCAN, DST, LIMS1, TNC, ASTN1, CTNND2, CTNND1, CDH2, NEO1, CDH4, CD24A, FAT3, PVRL3, TRO, TTYH1, MLLT4, LPP, NLGN1, PCDH19, LAMA1, ITGA9, CDH13, CDON, PSPC1 DCC, ENAH, PLXNA2, CAPZA2, ATP5B, ASTN1, PAX6, ZEB2, CDH2, CDH4, GLI3, CD24A, EPHB1, NRCAM, GO:0006928~cell CTTNBP2, EDNRB, APP, PTK2, ETV1, CLASP2, STRBP, 36 4.04 3.46E-07 510 205 7436 2.56 7.28E-04 3.64E-04 5.98E-04 motion NRG1, DCLK1, PLAT, SGPL1, TGFBR1, EVL, MYH9, YWHAE, NCKAP1, CTNNA2, SEMA6A, EPHA4, NDEL1, FYN, LRP6 PLXNA2, ADCY5, PAX6, GLI3, CTNNB1, LPHN2, EDNRB, LPHN3, APP, CSNK2A1, GPR45, NRG1, RAPGEF1, WWOX, SGPL1, TLE4, SPEN, NCAM1, DDR1, GRB10, GRM3, GNAQ, HIPK1, GNB1, HIPK2, PYGO1, GO:0007166~cell RNF138, ROR2, CNTN1, -
DLC1 Expression Is Reduced in Human Cutaneous Melanoma And
Modern Pathology (2014) 27, 1203–1211 & 2014 USCAP, Inc. All rights reserved 0893-3952/14 $32.00 1203 DLC1 expression is reduced in human cutaneous melanoma and correlates with patient survival Cecilia Sjoestroem1, Shahram Khosravi1, Yabin Cheng1, Gholamreza Safaee Ardekani1, Magdalena Martinka2 and Gang Li1 1Department of Dermatology and Skin Science, Vancouver Coastal Health Research Institute, University of British Columbia, Vancouver, BC, Canada and 2Department of Pathology, Vancouver General Hospital, Vancouver, BC, Canada Deleted in Liver Cancer-1 (DLC1) is a Rho-GTPase-activating protein known to be downregulated and function as a tumor suppressor in numerous solid and hematological cancers. Its expression status in melanoma is currently unknown however, prompting us to examine this. Using immunohistochemistry and tissue microarrays containing a large set of melanocytic lesions (n ¼ 539), we examined the expression profile of DLC1 in melanoma progression, as well as the association between DLC1 and patient survival. We detected both cytoplasmic and nuclear DLC1 expression, and found that whereas cytoplasmic DLC1 was significantly downregulated in metastatic melanoma compared with nevi and primary melanoma, nuclear DLC1 expression was significantly down in primary melanoma compared with nevi, and then further down in metastatic melanoma. Loss of cytoplasmic DLC1 was significantly associated with poorer overall and disease-specific 5-year survival rates of all melanoma (Po0.001 and P ¼ 0.001, respectively) and metastatic melanoma patients (P ¼ 0.020 and 0.008, respectively), and similar results were seen for nuclear DLC1 (Po0.001 for both overall and disease-specific survival for all melanoma patients, and P ¼ 0.004 for metastatic melanoma patients). -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Cooperative Antiproliferative Effect of Coordinated Ectopic Expression Of
ONCOLOGY LETTERS 12: 1591-1596, 2016 Cooperative antiproliferative effect of coordinated ectopic expression of DLC1 tumor suppressor protein and silencing of MYC oncogene expression in liver cancer cells: Therapeutic implications XUYU YANG1,2, XIAOLING ZHOU1,3, PAUL TONE4, MARIAN E. DURKIN1,5 and NICHOLAS C. POPESCU1,5 1Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health; 2Section on Cellular Neurobiology, Eunice Kennedy Shriver National Institute of Child Health and Development; 3Laboratory of Cancer Biology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 2089-4262; 4Department of Medicine, Richmond University Medical Center, Staten Island, NY 10310; 5Laboratory of Cellular Oncology, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892-4262, USA Received November 11, 2015; Accepted June 3, 2016 DOI: 10.3892/ol.2016.4781 Abstract. Human hepatocellular carcinoma (HCC) is one of the injection to mice bearing subcutaneous xenografts of Huh7 cells most common types of cancer and has a very poor prognosis; transfected with shMyc or control shRNA. A cooperative thus, the development of effective therapies for the treatment of inhibitory effect of DLC1 expression and c-Myc knockdown on advanced HCC is of high clinical priority. In the present study, the growth of Huh7-derived tumors was observed, suggesting the anti-oncogenic effect of combined knockdown of c-Myc that targeted liver cell delivery of DLC1 and c-Myc shRNA expression and ectopic restoration of deleted in liver cancer 1 may serve as a possible gene therapy modality for the treatment (DLC1) expression was investigated in human liver cancer of human HCC. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -

Identification of a Region of Homozygous Deletion on 8P22–23.1 in Medulloblastoma
Oncogene (2002) 21, 1461 ± 1468 ã 2002 Nature Publishing Group All rights reserved 0950 ± 9232/02 $25.00 www.nature.com/onc Identi®cation of a region of homozygous deletion on 8p22 ± 23.1 in medulloblastoma Xiao-lu Yin1, Jesse Chung-sean Pang1 and Ho-keung Ng*,1 1Department of Anatomical and Cellular Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, China To identify critical tumor suppressor loci that are radiotherapy and chemotherapy, have greatly improved associated with the development of medulloblastoma, the outcomes of patients. In the past 30 years, the 5- we performed a comprehensive genome-wide allelotype year survival rate has increased from 10 to about 50%. analysis in a series of 12 medulloblastomas. Non-random However, long-term survival in children with advanced allelic imbalances were identi®ed on chromosomes 7q disease is only about 30% (Giangaspero et al., 2000; (58.3%), 8p (66.7%), 16q (58.3%), 17p (58.3%) and Heideman et al., 1997). Further enhancement of 17q (66.7%). Comparative genomic hybridization analy- survival will rely on a better understanding of the sis con®rmed that allelic imbalances on 8p, 16q and 17p biology of this malignant disease to improve current were due to loss of genetic materials. Finer deletion treatments or to develop novel therapy. mapping in an expanded series of 23 medulloblastomas Non-random loss of genetic material from chromo- localized the common deletion region on 8p to an interval somal loci is a common feature in the development of of 18.14 cM on 8p22 ± 23.2.