Downloaded from Plntfdb ( To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalogue2013 Web.Pdf
bwfp British Wild Flower Plants www.wildflowers.co.uk Plants for Trade Plants for Home Specialist Species Wildflower Seed Green Roof Plants Over 350 species Scan here to of British native buy online plants 25th Anniversary Year Finding Us British Wild Flower Plants Burlingham Gardens 31 Main Road North Burlingham Norfolk NR13 4TA Phone / Fax: (01603) 716615 Email: [email protected] Website: http://www.wildflowers.co.uk Twitter: @WildflowersUK Nursery Opening Times Monday to Thursday: 10.00am - 4.00pm Friday: 10.00am - 2.30pm Please note that we are no longer open at weekends or Bank Holidays. Catalogue Contents Contact & Contents Page 02 About Us Page 03 Mixed Trays Pages 04-05 Reed Beds Page 06 Green Roofs Page 07 Wildflower Seeds Page 08 Planting Guide Pages 09-10 Attracting Wildlife Page 11 Rabbit-Proof Plants Page 12 List of Plants Pages 13-50 Scientific Name Look Up Pages 51-58 Terms & Conditions Page 59 www.wildflowers.co.uk 2 Tel/Fax:(01603)716615 About Us Welcome.... About Our Plants We are a family-run nursery, situated in Norfolk on a Our species are available most of the year in: six acre site. We currently stock over 350 species of 3 native plants and supply to all sectors of the industry Plugs: Young plants in 55cm cells with good rootstock. on a trade and retail basis. We are the largest grower of native plants in the UK and possibly Europe. Provenance Our species are drawn from either our own seed collections or from known provenance native sources. We comply with the Flora Locale Code of Practice. -

Review with Checklist of Fabaceae in the Herbarium of Iraq Natural History Museum
Review with checklist of Fabaceae in the herbarium of Iraq natural history museum Khansaa Rasheed Al-Joboury * Iraq Natural History Research Center and Museum, University of Baghdad, Baghdad, Iraq. GSC Biological and Pharmaceutical Sciences, 2021, 14(03), 137–142 Publication history: Received on 08 February 2021; revised on 10 March 2021; accepted on 12 March 2021 Article DOI: https://doi.org/10.30574/gscbps.2021.14.3.0074 Abstract This study aimed to make an inventory of leguminous plants for the purpose of identifying the plants that were collected over long periods and stored in the herbarium of Iraq Natural History Museum. It was found that the herbarium contains a large and varied number of plants from different parts of Iraq and in different and varied environments. It was collected and arranged according to a specific system in the herbarium to remain an important source for all graduate students and researchers to take advantage of these plants. Also, the flowering and fruiting periods of these plants in Iraq were recorded for different regions. Most of these plants begin to flower in the spring and thrive in fields and farms. Keywords: Fabaceae; Herbarium; Iraq; Natural; History; Museum 1. Introduction Leguminosae, Fabaceae or Papilionaceae, which was called as legume, pea, or bean Family, belong to the Order of Fabales [1]. The Fabaceae family have 727 genera also 19,325 species, which contents herbs, shrubs, trees, and climbers [2]. The distribution of fabaceae family was variety especially in cold mountainous regions for Europe, Asia and North America, It is also abundant in Central Asia and is characterized by great economic importance. -

Lessons from 20 Years of Plant Genome Sequencing: an Unprecedented Resource in Need of More Diverse Representation
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.31.446451; this version posted May 31, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Lessons from 20 years of plant genome sequencing: an unprecedented resource in need of more diverse representation Authors: Rose A. Marks1,2,3, Scott Hotaling4, Paul B. Frandsen5,6, and Robert VanBuren1,2 1. Department of Horticulture, Michigan State University, East Lansing, MI 48824, USA 2. Plant Resilience Institute, Michigan State University, East Lansing, MI 48824, USA 3. Department of Molecular and Cell Biology, University of Cape Town, Rondebosch 7701, South Africa 4. School of Biological Sciences, Washington State University, Pullman, WA, USA 5. Department of Plant and Wildlife Sciences, Brigham Young University, Provo, UT, USA 6. Data Science Lab, Smithsonian Institution, Washington, DC, USA Keywords: plants, embryophytes, genomics, colonialism, broadening participation Correspondence: Rose A. Marks, Department of Horticulture, Michigan State University, East Lansing, MI 48824, USA; Email: [email protected]; Phone: (603) 852-3190; ORCID iD: https://orcid.org/0000-0001-7102-5959 Abstract The field of plant genomics has grown rapidly in the past 20 years, leading to dramatic increases in both the quantity and quality of publicly available genomic resources. With an ever- expanding wealth of genomic data from an increasingly diverse set of taxa, unprecedented potential exists to better understand the evolution and genome biology of plants. -

State of New York City's Plants 2018
STATE OF NEW YORK CITY’S PLANTS 2018 Daniel Atha & Brian Boom © 2018 The New York Botanical Garden All rights reserved ISBN 978-0-89327-955-4 Center for Conservation Strategy The New York Botanical Garden 2900 Southern Boulevard Bronx, NY 10458 All photos NYBG staff Citation: Atha, D. and B. Boom. 2018. State of New York City’s Plants 2018. Center for Conservation Strategy. The New York Botanical Garden, Bronx, NY. 132 pp. STATE OF NEW YORK CITY’S PLANTS 2018 4 EXECUTIVE SUMMARY 6 INTRODUCTION 10 DOCUMENTING THE CITY’S PLANTS 10 The Flora of New York City 11 Rare Species 14 Focus on Specific Area 16 Botanical Spectacle: Summer Snow 18 CITIZEN SCIENCE 20 THREATS TO THE CITY’S PLANTS 24 NEW YORK STATE PROHIBITED AND REGULATED INVASIVE SPECIES FOUND IN NEW YORK CITY 26 LOOKING AHEAD 27 CONTRIBUTORS AND ACKNOWLEGMENTS 30 LITERATURE CITED 31 APPENDIX Checklist of the Spontaneous Vascular Plants of New York City 32 Ferns and Fern Allies 35 Gymnosperms 36 Nymphaeales and Magnoliids 37 Monocots 67 Dicots 3 EXECUTIVE SUMMARY This report, State of New York City’s Plants 2018, is the first rankings of rare, threatened, endangered, and extinct species of what is envisioned by the Center for Conservation Strategy known from New York City, and based on this compilation of The New York Botanical Garden as annual updates thirteen percent of the City’s flora is imperiled or extinct in New summarizing the status of the spontaneous plant species of the York City. five boroughs of New York City. This year’s report deals with the City’s vascular plants (ferns and fern allies, gymnosperms, We have begun the process of assessing conservation status and flowering plants), but in the future it is planned to phase in at the local level for all species. -
Species Lists
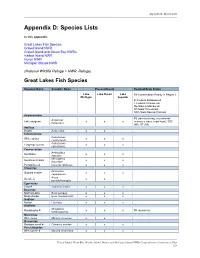
Appendix D: Species Lists Appendix D: Species Lists In this appendix: Great Lakes Fish Species Gravel Island NWR Gravel Island and Green Bay NWRs Harbor Island NWR Huron NWR Michigan Islands NWR (National Wildlife Refuge = NWR, Refuge) Great Lakes Fish Species Common Name Scientific Name Present/Absent Regional/State Status Lake Lake Huron Lake R3-Conservation Priority in Region 3 Michigan Superior E- Federal Endangered T-Federal Threatened SE-State Endangered ST-State Threatened SSC-State Special Concern Acipenseridae R3 (rare/declining, recreational/ Acipenser Lake sturgeon x x x economic value, tribal trust), SSC fulvescens (WI), ST (MI) Amiidae Bowfin Amia calva x x Catostomidae Catostomus White sucker x x x commersoni Catostomus Longnose sucker x x x catostomus Centrarchidae Ambloplites Rockbass x x x rupestris Micropterus Smallmouth bass x x x dolomieui Pumpkinseed Lepomis gibbosus x x x Clupeidae Dorosoma Gizzard shad # x x x cepedianum Alosa Alewife # x x pseudoharengus Cyprinidae Carp # Cyprinus Carpio x x x Esocidae Northern pike Esox Lucieus x x x Muskellunge Esox masquinongy x x x Gadidae Burbot Lota lota x x x Gobiidae Neogobius Round goby # x x x R3 (nuisance) melanostomus Moronidae White bass Morone chrysops x x Osmeridae Rainbow smelt # Osmerus mordax x x x Percichthyidae White perch # Morone americana x x x Gravel Island, Green Bay, Harbor Island, Huron, and Michigan Islands NWRs/Comprehensive Conservation Plan 221 Appendix D: Species Lists Common Name Scientific Name Present/Absent Regional/State Status Percidae R3 (rare/declining, -

Illinois Exotic Species List
Exotic Species in Illinois Descriptions for these exotic species in Illinois will be added to the Web page as time allows for their development. A name followed by an asterisk (*) indicates that a description for that species can currently be found on the Web site. This list does not currently name all of the exotic species in the state, but it does show many of them. It will be updated regularly with additional information. Microbes viral hemorrhagic septicemia Novirhabdovirus sp. West Nile virus Flavivirus sp. Zika virus Flavivirus sp. Fungi oak wilt Ceratocystis fagacearum chestnut blight Cryphonectria parasitica Dutch elm disease Ophiostoma novo-ulmi and Ophiostoma ulmi late blight Phytophthora infestans white-nose syndrome Pseudogymnoascus destructans butternut canker Sirococcus clavigignenti-juglandacearum Plants okra Abelmoschus esculentus velvet-leaf Abutilon theophrastii Amur maple* Acer ginnala Norway maple Acer platanoides sycamore maple Acer pseudoplatanus common yarrow* Achillea millefolium Japanese chaff flower Achyranthes japonica Russian knapweed Acroptilon repens climbing fumitory Adlumia fungosa jointed goat grass Aegilops cylindrica goutweed Aegopodium podagraria horse chestnut Aesculus hippocastanum fool’s parsley Aethusa cynapium crested wheat grass Agropyron cristatum wheat grass Agropyron desertorum corn cockle Agrostemma githago Rhode Island bent grass Agrostis capillaris tree-of-heaven* Ailanthus altissima slender hairgrass Aira caryophyllaea Geneva bugleweed Ajuga genevensis carpet bugleweed* Ajuga reptans mimosa -

Atlas of the Flora of New England: Fabaceae
Angelo, R. and D.E. Boufford. 2013. Atlas of the flora of New England: Fabaceae. Phytoneuron 2013-2: 1–15 + map pages 1– 21. Published 9 January 2013. ISSN 2153 733X ATLAS OF THE FLORA OF NEW ENGLAND: FABACEAE RAY ANGELO1 and DAVID E. BOUFFORD2 Harvard University Herbaria 22 Divinity Avenue Cambridge, Massachusetts 02138-2020 [email protected] [email protected] ABSTRACT Dot maps are provided to depict the distribution at the county level of the taxa of Magnoliophyta: Fabaceae growing outside of cultivation in the six New England states of the northeastern United States. The maps treat 172 taxa (species, subspecies, varieties, and hybrids, but not forms) based primarily on specimens in the major herbaria of Maine, New Hampshire, Vermont, Massachusetts, Rhode Island, and Connecticut, with most data derived from the holdings of the New England Botanical Club Herbarium (NEBC). Brief synonymy (to account for names used in standard manuals and floras for the area and on herbarium specimens), habitat, chromosome information, and common names are also provided. KEY WORDS: flora, New England, atlas, distribution, Fabaceae This article is the eleventh in a series (Angelo & Boufford 1996, 1998, 2000, 2007, 2010, 2011a, 2011b, 2012a, 2012b, 2012c) that presents the distributions of the vascular flora of New England in the form of dot distribution maps at the county level (Figure 1). Seven more articles are planned. The atlas is posted on the internet at http://neatlas.org, where it will be updated as new information becomes available. This project encompasses all vascular plants (lycophytes, pteridophytes and spermatophytes) at the rank of species, subspecies, and variety growing independent of cultivation in the six New England states. -

Biological Activities of Trifolium Pratense: a Review
Acta Scientific Pharmaceutical Sciences (ISSN: 2581-5423) Volume 3 Issue 9 September 2019 Review Article Biological Activities of Trifolium Pratense: A Review Atiq-ur-Rehman1,2* 1University College of Pharmacy, University of the Punjab, Lahore, Pakistan 2Faculty of Pharmacy, The University of Lahore, Lahore, Pakistan *Corresponding Author: Atiq-ur-Rehman, Faculty of Pharmacy, The University of Lahore and University College of Pharmacy, University of the Punjab, Lahore Pakistan. Received: July 25, 2019; Published: August 16, 2019 Abstract Trifolium pratense is an important plant of the Legume family. It has drawn the attention of several researchers around the globe. This plant was traditionally used as forage or as soil improver is now seen as the plant containing vast therapeutic activities which include anti-oxidative, anti-cancer, neuroprotective, anti-hyperglycemic, anti-hyperlipidemic, osteoprotective and cardio protective properties. The therapeutic properties are shown in various in vivo, in vitro and ex vivo experiments. The review highlights the Tri- forium pratense basic knowledge its extraction, components and their actions, major activities possessed by plant along with their mechanisms. Trifolium plant is mainmajorly used in menopausal women to reduce the discomfort and menopausal effects such as moderate cancer causing cells. Various strategies were applied and the plant is still under study for further development in its effects. hot flushes and increase in breast density. The plant is also majorly responsible for preventing breast cancer and other apoptosis of Keywords: Trifolium Pratense; Cancer; Trifolium Introduction Family The genus Trifolium comprises of almost 240 species each re- It belongs to the family Fabeaceae leguminosae. markable for its agricultural and therapeutic effects. -

RED CLOVER This Plant May Become Weedy Or Invasive in Some Regions Or Habitats and May Displace Other Vegetation Trifolium Pratense L
Plant Guide Weediness RED CLOVER This plant may become weedy or invasive in some regions or habitats and may displace other vegetation Trifolium pratense L. if not properly managed. Consult your local NRCS Plant Symbol = TRPR2 Field Office, Cooperative Extension Service Office, or state natural resource or agriculture department Contributed by: Idaho Plant Materials regarding its status and use. Weed information is also available from the PLANTS web site at plants.usda.gov Description Trifolium pratense L., red clover, is an introduced biennial or short-lived perennial that grows as one of two types: medium (double-cut) or mammoth (single- cut). Red clover initiates growth from the plant crown. Plants have hollow, hairy stems and branches. Stem lengths of medium and mammoth types average 18 inches and 24-30 inches, respectively. Medium types have about 4 branches per stem; mammoth types have 6 branches per stem. USDA NRCS PLANTS Each leaf consists of a slender stalk bearing 3 leaflets. The taproot of red clover normally disintegrates in the second year and plants that Uses survive have developed secondary, extensively Red clover is the most widely grown of all the true branched roots. Flowers are borne in compact clovers and is the most important legume hay crop in clusters at the tips of the branches and are usually the northeastern United States. Red clover is rose-pink in color. The flower shape is similar to pea primarily used for hay, pasture, silage, and soil flowers except is more elongated and much smaller. improvement. It is a quick growing crop, easily Flower heads usually consist of up to 125 flowers. -

Conservation Plants Pocket ID Guide
About this guide.......... The purpose of this guide is to help you identify come commonly used conservation plants. Its color photos, line drawings and seed photos will help you make identifications. Also included are plant stand evaluation and recommended use charts. Keep this guide with you as long as you need it! Uses 1 2 Evaluating Stands Seeding success may not be obvious from visual observation. Use the chart below to determine whether your first-year stand is adequate. Lay a square-foot frame, or a circular frame with a 42.5-inch circumference on the ground. Count the number of seedlings within the frame, taking at least 10 counts for each 10 acres, in representative areas of the field. The table is based on pure stands; if a mixture of grass and legume is planted, reduce the numbers by the ratio of each species planted. Inadequate stands should be re-seeded. When a stand is judged to be between adequate and inadequate, it should be reevaluated after the second growing season. Warm-season grasses may need to be evaluated after the third growing season. 3 Seeds Early ID -- Seed is Key Grasses can be very difficult to identify in early growth stages. The seed may be the best identify aid. A seed retains its form and position in the ground through the seedling's early growth stages. To identify a seedling, carefully dig it up and compare it to photographs or actual seeds. 4 Plant Parts Conservation Plants The Elsberry Plant Materials Center The Elsberry Plant Materials Center is a 243-acre facility near Elsberry, Missouri. -

Comparative Analysis of Nodulation-Related Small Secreted Peptides Across Legume Species
Comparative analysis of nodulation-related small secreted peptides across legume species A DISSERTATION SUBMITTED TO THE FACULTY OF THE UNIVERSITY OF MINNESOTA BY Diana Trujillo IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Nevin Young November 2017 © Diana Trujillo 2017 Acknowledgments I would like to thank all those who, one way or another, have made this work possible. First and foremost, thanks go to my advisor, Nevin Young, who helped shape my vision for this project and was always available to give me support or the necessary guidance in the right direction. I would also like to thank Kevin Silverstein who played a large role during the development of my LSE pipeline, and was a helpful mentor in bioinformatic matters. Thanks to the other members of my advisory committee, Peter Morrell, Michael Sadowsky, and Robert Stupar, who provided a fresh outlook and valuable advice that helped to improve this study. I would like to thank Joseph Guhlin and Peng Zhou who were always one step away when I had Unix concerns, and Shaun Curtin and Roxanne Denny who guided me through the technical aspects of growing or transforming Medicago. I am grateful that I had a strong network of friends and colleagues to discuss biology, coding, and life. To Allison Haaning, Beth Fallon, Christina Smith, Leland Werden, Derek Nedveck, and Eli Krumholz: thanks for sharing the journey. I would like to thank my mother, María Cecilia, who instilled my love of reading, truth-seeking, and plants. To my husband, Dylan Huss, thank you for always calling to see if I already ate (including as I typed these words). -

Biology of a New Virus Isolated from Lupinus Nootkatensis Plants in Alaska
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln U.S. Department of Agriculture: Agricultural Publications from USDA-ARS / UNL Faculty Research Service, Lincoln, Nebraska 2004 Biology of a new virus isolated from Lupinus nootkatensis plants in Alaska N. L. Robertson USDA, ARS Follow this and additional works at: https://digitalcommons.unl.edu/usdaarsfacpub Robertson, N. L., "Biology of a new virus isolated from Lupinus nootkatensis plants in Alaska" (2004). Publications from USDA-ARS / UNL Faculty. 1443. https://digitalcommons.unl.edu/usdaarsfacpub/1443 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Agricultural Research Service, Lincoln, Nebraska at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Publications from USDA-ARS / UNL Faculty by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Plant Pathology (2004) 53, 569–576 Doi: 10.1046/j.0032-0862.2004.01058.x BlackwellBiology Publishing, Ltd. of a new virus isolated from Lupinus nootkatensis plants in Alaska N. L. Robertson USDA, ARS, Arctic Plant Germplasm Introduction and Research Project, 533 E. Fireweed Avenue, Palmer, AK 99645, USA A new virus named Nootka lupine vein-clearing virus (NLVCV) was isolated from Lupinus nootkatensis plants that were confined to a relatively small area in the Talkeetna mountains of south-central Alaska. Annual surveys (2000–03) con- sistently found leaf symptoms of pronounced vein clearing and mosaic on 3- to 4-week-old plants in late June. Spherical particles ≈30 nm in diameter were isolated from these leaves. Virions contained a single-stranded RNA of ≈4·0–4·2 kb and one species of capsid protein estimated to be ≈40 kDa.