Comparative Analysis of Nodulation-Related Small Secreted Peptides Across Legume Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nutrition Facts N *Learn About Fiber on Page 2
Health and Learning Success Go Hand-In-Hand California’s geography offers a bounty of fresh produce and recreational areas. From stone fruits and salad greens to state and local parks, there is no shortage of healthy foods to eat and outdoor activities to do in California. Studies show that healthy eating and physical activity are correlated with improved academic achievement. Use Harvest of the Month to allow students to experience California-grown fruit and vegetables with their senses. Teach students to live a healthy, active lifestyle and support academic content standards to link the classroom, cafeteria, home, and community. Exploring California Peaches: Taste Testing Network for a Healthy California What You Will Need (per group of 8 students): n Four ripe peaches and four ripe nectarines (two each of yellow and white varieties)* n Paring knife and cutting board n Paper towels *Choose peaches and nectarines that are fragrant and firm to slightly soft when pressed. Optional: Sample other stone fruits (cherries, plums, apricots, etc.) with peaches. Activity: n Distribute yellow peaches and nectarines to each student group. n Observe the look, feel, and smell of each; record observations. n Cut open the second yellow fruit; observe the taste and sound and record observations. n Repeat with white peaches and nectarines. n Discuss similarities and differences among the four varieties. n Record students’ favorite variety; share results with school nutrition staff. For more ideas, reference: Nutrition to Grow On, CDE, 2001. Cooking in Class: Reasons to Eat Peaches Peach Smoothies A ½ cup of sliced peaches (about half of Makes 35 tastes at ¼ cup each a medium peach) provides: Ingredients: n A source of vitamin A and vitamin C. -

State of New York City's Plants 2018
STATE OF NEW YORK CITY’S PLANTS 2018 Daniel Atha & Brian Boom © 2018 The New York Botanical Garden All rights reserved ISBN 978-0-89327-955-4 Center for Conservation Strategy The New York Botanical Garden 2900 Southern Boulevard Bronx, NY 10458 All photos NYBG staff Citation: Atha, D. and B. Boom. 2018. State of New York City’s Plants 2018. Center for Conservation Strategy. The New York Botanical Garden, Bronx, NY. 132 pp. STATE OF NEW YORK CITY’S PLANTS 2018 4 EXECUTIVE SUMMARY 6 INTRODUCTION 10 DOCUMENTING THE CITY’S PLANTS 10 The Flora of New York City 11 Rare Species 14 Focus on Specific Area 16 Botanical Spectacle: Summer Snow 18 CITIZEN SCIENCE 20 THREATS TO THE CITY’S PLANTS 24 NEW YORK STATE PROHIBITED AND REGULATED INVASIVE SPECIES FOUND IN NEW YORK CITY 26 LOOKING AHEAD 27 CONTRIBUTORS AND ACKNOWLEGMENTS 30 LITERATURE CITED 31 APPENDIX Checklist of the Spontaneous Vascular Plants of New York City 32 Ferns and Fern Allies 35 Gymnosperms 36 Nymphaeales and Magnoliids 37 Monocots 67 Dicots 3 EXECUTIVE SUMMARY This report, State of New York City’s Plants 2018, is the first rankings of rare, threatened, endangered, and extinct species of what is envisioned by the Center for Conservation Strategy known from New York City, and based on this compilation of The New York Botanical Garden as annual updates thirteen percent of the City’s flora is imperiled or extinct in New summarizing the status of the spontaneous plant species of the York City. five boroughs of New York City. This year’s report deals with the City’s vascular plants (ferns and fern allies, gymnosperms, We have begun the process of assessing conservation status and flowering plants), but in the future it is planned to phase in at the local level for all species. -
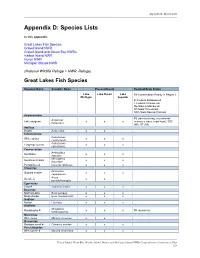
Species Lists
Appendix D: Species Lists Appendix D: Species Lists In this appendix: Great Lakes Fish Species Gravel Island NWR Gravel Island and Green Bay NWRs Harbor Island NWR Huron NWR Michigan Islands NWR (National Wildlife Refuge = NWR, Refuge) Great Lakes Fish Species Common Name Scientific Name Present/Absent Regional/State Status Lake Lake Huron Lake R3-Conservation Priority in Region 3 Michigan Superior E- Federal Endangered T-Federal Threatened SE-State Endangered ST-State Threatened SSC-State Special Concern Acipenseridae R3 (rare/declining, recreational/ Acipenser Lake sturgeon x x x economic value, tribal trust), SSC fulvescens (WI), ST (MI) Amiidae Bowfin Amia calva x x Catostomidae Catostomus White sucker x x x commersoni Catostomus Longnose sucker x x x catostomus Centrarchidae Ambloplites Rockbass x x x rupestris Micropterus Smallmouth bass x x x dolomieui Pumpkinseed Lepomis gibbosus x x x Clupeidae Dorosoma Gizzard shad # x x x cepedianum Alosa Alewife # x x pseudoharengus Cyprinidae Carp # Cyprinus Carpio x x x Esocidae Northern pike Esox Lucieus x x x Muskellunge Esox masquinongy x x x Gadidae Burbot Lota lota x x x Gobiidae Neogobius Round goby # x x x R3 (nuisance) melanostomus Moronidae White bass Morone chrysops x x Osmeridae Rainbow smelt # Osmerus mordax x x x Percichthyidae White perch # Morone americana x x x Gravel Island, Green Bay, Harbor Island, Huron, and Michigan Islands NWRs/Comprehensive Conservation Plan 221 Appendix D: Species Lists Common Name Scientific Name Present/Absent Regional/State Status Percidae R3 (rare/declining, -

Illinois Exotic Species List
Exotic Species in Illinois Descriptions for these exotic species in Illinois will be added to the Web page as time allows for their development. A name followed by an asterisk (*) indicates that a description for that species can currently be found on the Web site. This list does not currently name all of the exotic species in the state, but it does show many of them. It will be updated regularly with additional information. Microbes viral hemorrhagic septicemia Novirhabdovirus sp. West Nile virus Flavivirus sp. Zika virus Flavivirus sp. Fungi oak wilt Ceratocystis fagacearum chestnut blight Cryphonectria parasitica Dutch elm disease Ophiostoma novo-ulmi and Ophiostoma ulmi late blight Phytophthora infestans white-nose syndrome Pseudogymnoascus destructans butternut canker Sirococcus clavigignenti-juglandacearum Plants okra Abelmoschus esculentus velvet-leaf Abutilon theophrastii Amur maple* Acer ginnala Norway maple Acer platanoides sycamore maple Acer pseudoplatanus common yarrow* Achillea millefolium Japanese chaff flower Achyranthes japonica Russian knapweed Acroptilon repens climbing fumitory Adlumia fungosa jointed goat grass Aegilops cylindrica goutweed Aegopodium podagraria horse chestnut Aesculus hippocastanum fool’s parsley Aethusa cynapium crested wheat grass Agropyron cristatum wheat grass Agropyron desertorum corn cockle Agrostemma githago Rhode Island bent grass Agrostis capillaris tree-of-heaven* Ailanthus altissima slender hairgrass Aira caryophyllaea Geneva bugleweed Ajuga genevensis carpet bugleweed* Ajuga reptans mimosa -

Biological Activities of Trifolium Pratense: a Review
Acta Scientific Pharmaceutical Sciences (ISSN: 2581-5423) Volume 3 Issue 9 September 2019 Review Article Biological Activities of Trifolium Pratense: A Review Atiq-ur-Rehman1,2* 1University College of Pharmacy, University of the Punjab, Lahore, Pakistan 2Faculty of Pharmacy, The University of Lahore, Lahore, Pakistan *Corresponding Author: Atiq-ur-Rehman, Faculty of Pharmacy, The University of Lahore and University College of Pharmacy, University of the Punjab, Lahore Pakistan. Received: July 25, 2019; Published: August 16, 2019 Abstract Trifolium pratense is an important plant of the Legume family. It has drawn the attention of several researchers around the globe. This plant was traditionally used as forage or as soil improver is now seen as the plant containing vast therapeutic activities which include anti-oxidative, anti-cancer, neuroprotective, anti-hyperglycemic, anti-hyperlipidemic, osteoprotective and cardio protective properties. The therapeutic properties are shown in various in vivo, in vitro and ex vivo experiments. The review highlights the Tri- forium pratense basic knowledge its extraction, components and their actions, major activities possessed by plant along with their mechanisms. Trifolium plant is mainmajorly used in menopausal women to reduce the discomfort and menopausal effects such as moderate cancer causing cells. Various strategies were applied and the plant is still under study for further development in its effects. hot flushes and increase in breast density. The plant is also majorly responsible for preventing breast cancer and other apoptosis of Keywords: Trifolium Pratense; Cancer; Trifolium Introduction Family The genus Trifolium comprises of almost 240 species each re- It belongs to the family Fabeaceae leguminosae. markable for its agricultural and therapeutic effects. -

Peony, Powdery Mildew (Erysiphe Polygoni)
Problem: Peony, Powdery Mildew (Erysiphe polygoni) Host Plants: Peony Description: Powdery mildew starts as individual spots that resemble snowflakes but rapidly coalesce to cover the entire leaf so that a plant look like it was dusted with flour. Though common on other plants such as lilac and bee balm, it has been relatively rare on peony until the last few years. Recommendations: Poor air movement and shade make the disease more likely. Growing peonies in full sun with good air movement will help minimize the disease. Fungicides can be effective if applied before infection has occurred. Therefore, heavily infected plants should not be treated as the treatment will be ineffective. Fortunately, the disease should cause no lasting damage to the plant. Remove and discard (or compost) infected plant material at the end of the season. Look for individual spots to appear the following spring and then apply a recommended fungicide before the disease has spread. Suggested fungicides include myclobutanil (Eagle, Spectracide Immunox, Monterey Fungi-Maxx, Fertilome F- Stop Lawn & Garden Fungicide), propiconazole (Banner MAXX, Fertilome Liquid Systemic Fungicide, Bonide Infuse Systemic Disease Control) or tebuconazole (BioAdvanced Disease Control for Roses, Flowers & Shrubs). References: 1. Powdery Mildew in the Flower Garden, University of Minnesota Extension 2. Peony Powdery Mildew, University of Illinois Extension, Home, Yard & Garden Pest Newsletter, July 16, 2010 Last Update: 1/16/2020 Brand names appearing in this publication are for product identification purposes only. No endorsement is intended, nor is criticism implied of similar products not mentioned. Kansas State University Agricultural Experiment Station and Cooperative Extension Service . -

Peach Volatile Emission and Attractiveness of Different Host Plant
www.nature.com/scientificreports OPEN Peach volatile emission and attractiveness of diferent host plant volatiles blends to Cydia molesta in adjacent peach and pear orchards Peng‑fei Lu1* & Hai‑li Qiao2 The oriental fruit moth (OFM), Cydia (= Grapholitha) molesta, is a highly damaging pest; peaches are its primary host, and pears serve as post‑peach secondary hosts during the late season in China. We collected volatiles from detached peach shoots and fruits, and identifed them with gas chromatography–mass spectrometry (GC–MS). Antennally active compounds were identifed by gas chromatography-electroantennogram detection (GC-EAD), and these were further tested in the laboratory and feld. We detected consistent electroantennographic activity was for ten compounds. Signifcantly more C. molesta females were caught with a mixture of female EAD-active compounds identifed from the detached matured peach fruits (nonanal, butyl acetate, 3-methylbutyl acetate, hexyl acetate, (Z)-3-hexenyl acetate, linalool and farnesene) than other mixtures mimicking the volatile profle from detached matured fruits or shoots. We identifed a new GC-EAD active mixture from intact peach shoots composed of nonanal, (Z)-3-hexenyl acetate, (E)-β-ocimene, and 6-methyl- 5-hepten-2-one. In the feld test, the background odour of orchards could afect trap catches, and two peach-derived blends together with two previously known pear-derived blends were proven to be able to monitor the seasonal OFM population dispersal in adjacent orchards. These host plant blends will be efective for further designing candidate attractants for season-long C. molesta population dynamic monitoring. Phytochemicals are important olfactory cues for moths to fnd hosts and lay eggs 1–4. -

French Toast Crunch Kale + Bacon Hash Brown Casserole
SEASON 2 | EPISODE 01 and slowly stir, until sugar dissolves, sheet pan with a cooling rack on top of French Toast using a wooden spoon or heat resistant it. This will keep the toast from getting spatula. You’re looking for the mixture soggy while you cook the rest of your Crunch to become an amber color. slices. Repeat until all bread is cooked. prep: 1 hour cook: 10 minutes 4. Immediately take the mixture off the 4. Diagonally cut each slice of french cool: 10 minutes heat and stir in your butter, once butter toast, so that it makes 2 halves out of 1 melts, slowly stir in the heavy cream slice of bread. and salt. caramel crunch 5. Place 5 halves of french toast on a 4 cups corn flakes 5. Grease a sheet pan and set aside. plate and sprinkle with caramel crunch 3/4 cup sugar that was set aside and powdered sugar. 6. Add corn flakes into a large mixing 1/8 teaspoon cream of tartar bowl. Gently stir in the caramel sauce Top with syrup, sliced strawberries, 2 tablespoons water and cinnamon. Toss until cornflakes and a dollop of whipped cream. Serve 3 tablespoons butter, unsalted are coated, and spread into a single immediately. 1/4 cup heavy cream layer on a greased sheet pan. Allow this Makes 6-8 servings Pinch of salt to cool for about an hour 11/2 teaspoon cinnamon 7. Set a small amount of your crunchy mixture to the side for garnish, about 2 french toast cups. Take the remaining mixture and Kale + Bacon 12 slices of Brioche put into a plastic bag, leaving a small gap for air so as to not break the bag. -

Peach Fruit Fly, Bactrocera Zonata, Host List the Berries, Fruit, Nuts and Vegetables of the Listed Plant Species Are Now Considered Host Articles for B
June 2017 Peach fruit fly, Bactrocera zonata, Host List The berries, fruit, nuts and vegetables of the listed plant species are now considered host articles for B. zonata. Unless proven otherwise, all cultivars, varieties, and hybrids of the plant species listed herein are considered suitable hosts of B. zonata. Scientific Name Common Name Aegle marmelos (L.) Correa Baeltree Afzelia xylocarpa (Kurz) Craib Doussie Annona reticulata L. Custard apple Annona squamosa L. Sugar apple Careya arborea Roxb. Patana oak Carica papaya L. Papaya Casimiroa edulis La Llave & Lex. White sapote Citrullus lanatus (Thunb.) Matsum. & Nakai Watermelon Citrus aurantium L. Sour orange Citrus limon (L.) Burm. f. Lemon Citrus nobilis x Citrus deliciosa Ten. Kinnow Citrus paradisi Macfad. Grapefruit Citrus reticulata Blanco Mandarin orange Citrus sinensis (L.) Osbeck Orange Coccinia grandis (L.) Voigt Ivy gourd Cucumis sativus L. Cucumber Cydonia oblonga Mill. Quince Elaeocarpus hygrophilus Kurz Ma-kok-nam Eriobotrya japonica (Thunb.) Lindl. Loquat Ficus carica L. Fig Grewia asiatica L. Phalsa Lagenaria siceraria (Molina) Standl. Bottle gourd Luffa acutangula (L.) Roxb. Angled loofah Luffa spp. Tori Malpighia emarginata DC. Barbados cherry Malus pumila Mill. Apple Malus sylvestris (L.) Mill. Crab apple Mangifera indica L. Mango Manilkara zapota (L.) P. Royen Sapodilla Mimusops elengi L. Spanish cherry Momordica charantia L. Bitter melon Olea europaea L. Olive Persea americana Mill. Avocado Phoenix dactylifera L. Date palm Prunus armeniaca L. Apricot Prunus avium (L.) L. Sweet cherry Prunus domestica L. Plum Prunus persica (L.) Batsch Peach Prunus sp. N/A Psidium cattleyanum Sabine Strawberry guava Psidium guajava L. Guava Punica granatum L. Pomegranate Putranjiva roxburghii Wall. -

RED CLOVER This Plant May Become Weedy Or Invasive in Some Regions Or Habitats and May Displace Other Vegetation Trifolium Pratense L
Plant Guide Weediness RED CLOVER This plant may become weedy or invasive in some regions or habitats and may displace other vegetation Trifolium pratense L. if not properly managed. Consult your local NRCS Plant Symbol = TRPR2 Field Office, Cooperative Extension Service Office, or state natural resource or agriculture department Contributed by: Idaho Plant Materials regarding its status and use. Weed information is also available from the PLANTS web site at plants.usda.gov Description Trifolium pratense L., red clover, is an introduced biennial or short-lived perennial that grows as one of two types: medium (double-cut) or mammoth (single- cut). Red clover initiates growth from the plant crown. Plants have hollow, hairy stems and branches. Stem lengths of medium and mammoth types average 18 inches and 24-30 inches, respectively. Medium types have about 4 branches per stem; mammoth types have 6 branches per stem. USDA NRCS PLANTS Each leaf consists of a slender stalk bearing 3 leaflets. The taproot of red clover normally disintegrates in the second year and plants that Uses survive have developed secondary, extensively Red clover is the most widely grown of all the true branched roots. Flowers are borne in compact clovers and is the most important legume hay crop in clusters at the tips of the branches and are usually the northeastern United States. Red clover is rose-pink in color. The flower shape is similar to pea primarily used for hay, pasture, silage, and soil flowers except is more elongated and much smaller. improvement. It is a quick growing crop, easily Flower heads usually consist of up to 125 flowers. -

Conservation Plants Pocket ID Guide
About this guide.......... The purpose of this guide is to help you identify come commonly used conservation plants. Its color photos, line drawings and seed photos will help you make identifications. Also included are plant stand evaluation and recommended use charts. Keep this guide with you as long as you need it! Uses 1 2 Evaluating Stands Seeding success may not be obvious from visual observation. Use the chart below to determine whether your first-year stand is adequate. Lay a square-foot frame, or a circular frame with a 42.5-inch circumference on the ground. Count the number of seedlings within the frame, taking at least 10 counts for each 10 acres, in representative areas of the field. The table is based on pure stands; if a mixture of grass and legume is planted, reduce the numbers by the ratio of each species planted. Inadequate stands should be re-seeded. When a stand is judged to be between adequate and inadequate, it should be reevaluated after the second growing season. Warm-season grasses may need to be evaluated after the third growing season. 3 Seeds Early ID -- Seed is Key Grasses can be very difficult to identify in early growth stages. The seed may be the best identify aid. A seed retains its form and position in the ground through the seedling's early growth stages. To identify a seedling, carefully dig it up and compare it to photographs or actual seeds. 4 Plant Parts Conservation Plants The Elsberry Plant Materials Center The Elsberry Plant Materials Center is a 243-acre facility near Elsberry, Missouri. -

Biology of a New Virus Isolated from Lupinus Nootkatensis Plants in Alaska
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln U.S. Department of Agriculture: Agricultural Publications from USDA-ARS / UNL Faculty Research Service, Lincoln, Nebraska 2004 Biology of a new virus isolated from Lupinus nootkatensis plants in Alaska N. L. Robertson USDA, ARS Follow this and additional works at: https://digitalcommons.unl.edu/usdaarsfacpub Robertson, N. L., "Biology of a new virus isolated from Lupinus nootkatensis plants in Alaska" (2004). Publications from USDA-ARS / UNL Faculty. 1443. https://digitalcommons.unl.edu/usdaarsfacpub/1443 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Agricultural Research Service, Lincoln, Nebraska at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Publications from USDA-ARS / UNL Faculty by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Plant Pathology (2004) 53, 569–576 Doi: 10.1046/j.0032-0862.2004.01058.x BlackwellBiology Publishing, Ltd. of a new virus isolated from Lupinus nootkatensis plants in Alaska N. L. Robertson USDA, ARS, Arctic Plant Germplasm Introduction and Research Project, 533 E. Fireweed Avenue, Palmer, AK 99645, USA A new virus named Nootka lupine vein-clearing virus (NLVCV) was isolated from Lupinus nootkatensis plants that were confined to a relatively small area in the Talkeetna mountains of south-central Alaska. Annual surveys (2000–03) con- sistently found leaf symptoms of pronounced vein clearing and mosaic on 3- to 4-week-old plants in late June. Spherical particles ≈30 nm in diameter were isolated from these leaves. Virions contained a single-stranded RNA of ≈4·0–4·2 kb and one species of capsid protein estimated to be ≈40 kDa.