General Base-General Acid Catalysis by Terpenoid Cyclases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rehmannia Glutinosa-Monocultured Rhizosphere Soil
Comparative Metaproteomic Analysis on Consecutively Rehmannia glutinosa-Monocultured Rhizosphere Soil Linkun Wu1,2, Haibin Wang1,2., Zhixing Zhang1,2., Rui Lin2,3, Zhongyi Zhang1,4, Wenxiong Lin1,2* 1 School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2 Agroecological Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 3 College of Oceanography and Environmental Science, Xiamen University, Xiamen, Fujian, China, 4 Institute of Chinese Medicinal Materials, Henan Agriculture University, Zhengzhou, Henan, China Abstract Background: The consecutive monoculture for most of medicinal plants, such as Rehmannia glutinosa, results in a significant reduction in the yield and quality. There is an urgent need to study for the sustainable development of Chinese herbaceous medicine. Methodology/Principal Findings: Comparative metaproteomics of rhizosphere soil was developed and used to analyze the underlying mechanism of the consecutive monoculture problems of R. glutinosa. The 2D-gel patterns of protein spots for the soil samples showed a strong matrix dependency. Among the spots, 103 spots with high resolution and repeatability were randomly selected and successfully identified by MALDI TOF-TOF MS for a rhizosphere soil metaproteomic profile analysis. These proteins originating from plants and microorganisms play important roles in nutrient cycles and energy flow in rhizospheric soil ecosystem. They function in protein, nucleotide and secondary metabolisms, signal transduction and resistance. Comparative metaproteomics analysis revealed 33 differentially expressed protein spots in rhizosphere soil in response to increasing years of monoculture. Among them, plant proteins related to carbon and nitrogen metabolism and stress response, were mostly up-regulated except a down-regulated protein (glutathione S-transferase) involving detoxification. -
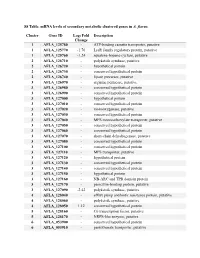
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Electric Supporting Information (ESI) Crystal Structure and Functional
Electronic Supplementary Material (ESI) for Chemical Science. This journal is © The Royal Society of Chemistry 2018 Electric Supporting Information (ESI) Crystal structure and functional analysis of large-terpene synthase belonging to a newly found subclass Masahiro Fujihashi,a* Tsutomu Sato,b* Yuma Tanaka,a Daisuke Yamamoto,a Tomoyuki Nishi,b Daijiro Ueda,b Mizuki Murakami,b Yoko Yasuno,c Ai Sekihara,c Kazuma Fuku,c Tetsuro Shinadac & Kunio Mikia* a. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: [email protected], [email protected] b. Department of Applied Biological Chemistry, Faculty of Agriculture, and Graduate School of Science and Technology, Niigata University, 8050 Ikarashi-2, Niigata 950-2181, Japan. E-mail: [email protected] c. Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. S1 Table of Contents Experimental Procedures S3 General procedure S3 Vector construction, expression and purification of BalTS S3 Crystallization and crystallographic analysis of balTS S3 Oligomeric state analysis S4 Analysis of the dimer geometry S4 Vector construction, expression and purification of BsuTS S4 Homology modeling of BsuTS S4 Enzymatic assays S5 Isolation and identification of β-springen S5 Movie Captions S6 Author Contributions S6 Figures Fig. S1 S7 Fig. S2 S8 Fig. S3 S9 Fig. S4 S10 Fig. S5 S17 Fig. S6 S18 Fig. S7, S8 S19 Fig. S9 S20 Fig. S10 S21 Fig. S11 S22 Tables Table S1 S23 Table S2 S24 References S27 S2 Experimental Procedures General procedure NMR spectra were recorded using a Bruker DPX 400 spectrometer at 400 MHz for proton (1H) and 100 MHz for carbon (13C). -

Hydroxylation of 1-Deoxypentalenic Acid Mediated by CYP105D7 (SAV 7469) of Streptomyces Avermitilis
The Journal of Antibiotics (2011) 64, 65–71 & 2011 Japan Antibiotics Research Association All rights reserved 0021-8820/11 $32.00 www.nature.com/ja ORIGINAL ARTICLE Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis Satoshi Takamatsu1,4, Lian-Hua Xu2,4, Shinya Fushinobu2, Hirofumi Shoun2, Mamoru Komatsu1, David E Cane3 and Haruo Ikeda1 Pentalenic acid (1) has been isolated from many Streptomyces sp. as a co-metabolite of the sesquiterpenoid antibiotic pentalenolactone and related natural products. We have previously reported the identification of a 13.4-kb gene cluster in the genome of Streptomyces avermitilis implicated in the biosynthesis of the pentalenolactone family of metabolites consisting of 13 open reading frames. Detailed molecular genetic and biochemical studies have revealed that at least seven genes are involved in the biosynthesis of the newly discovered metabolites, neopentalenoketolactone, but no gene specifically dedicated to the formation of pentalenic acid (1) was evident in the same gene cluster. The wild-type strain of S. avermitilis, as well as its derivatives, mainly produce pentalenic acid (1), together with neopentalenoketolactone (9). Disruption of the sav7469 gene encoding a cytochrome P450 (CYP105D7), members of which class are associated with the hydroxylation of many structurally different compounds, abolished the production of pentalenic acid (1). The sav7469-deletion mutant derived from SUKA11 carrying pKU462Hptl-clusterDptlH accumulated 1-deoxypentalenic acid (5), but not pentalenic acid (1). Reintroduction of an extra-copy of the sav7469 gene to SUKA11 Dsav7469 carrying pKU462Hptl-clusterDptlH restored the production of pentalenic acid (1). -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Diene Synthase for the Production of Novel Products
Engineering Selina-4(15),7(11)-diene synthase for the Production of Novel Products Emily Turri Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds Astbury Centre for Structural Molecular Biology September 2020 The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Emily Turri to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2020 The University of Leeds and Emily Turri 2 Acknowledgements Throughout my PhD I have been extremely lucky to receive support and encouragement from family, friends, colleagues and the university. Without these people and their support, this thesis would not be possible. First, I would like to thank my supervisors Alan Berry and Adam Nelson for their encouragement, guidance and patience over these four years. I would also like to thank Glyn Hemsworth and Nasir Khan for their continued advice and support. I am very grateful to Glyn Hemsworth, Sheena Radford, David Brockwell and John Blacker for letting me use their equipment while offering me direction to give me the best results. In addition, I would like to thank James Ault, Rachel George, Mary Bayana, Mark Howard and Ricardo Labes for their technical assistance and biochemical analysis. My PhD experience would have been nothing without the present and past members of the Hemrry’s and Radford’s with specific thanks to Jess, Alex, Jess and Dan. -

(12) Patent Application Publication (10) Pub. No.: US 2004/0229367 A1 Berka Et Al
US 2004O229367A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0229367 A1 Berka et al. (43) Pub. Date: Nov. 18, 2004 (54) METHODS FOR MONITORING MULTIPLE Related U.S. Application Data GENE EXPRESSION (60) Division of application No. 09/533,559, filed on Mar. (75) Inventors: Randy M. Berka, Davis, CA (US); 22, 2000, which is a continuation-in-part of applica Michael W. Rey, Davis, CA (US); tion No. 09/273,623, filed on Mar. 22, 1999, now Jeffrey R. Shuster, Davis, CA (US); abandoned. Sakari Kauppinen, Smoerum (DK); Ib Groth Clausen, Hillerod (DK); Peter Publication Classification Bjarke Olsen, Copenhagen (DK) 51)1) Int. Cl.C.7 ............................. C12N 15/745/74; C12N 1/16 Correspondence Address: (52) U.S. Cl. ......................................... 435/.484; 435/254.3 NOVOZYMES BIOTECH, INC. (57) ABSTRACT 1445. DREWAVE The present invention relates to methods for monitoring DAVIS, CA 95616 (US) differential expression of a plurality of genes in a first filamentous fungal cell relative to expression of the same genes in one or more Second filamentous fungal cells using (73) Assignees: Novozymes Biotech, Inc., Davis, CA; microarrays containing filamentous fungal expressed Novozymes A/S, Inc., Bagsvaerd (DK) Sequenced tags. The present invention also relates to fila mentous fungal expressed Sequenced tags and to computer (21) Appl. No.: 10/653,047 readable media and Substrates containing Such expressed Sequenced tags for monitoring expression of a plurality of (22) Filed: Aug. 29, 2003 genes in filamentous fungal cells. US 2004/0229367 A1 Nov. 18, 2004 METHODS FOR MONITORING MULTIPLE GENE organisms whose genomes have not been Sequenced. -

Computational-Guided Discovery and Characterization of a Sesquiterpene Synthase from Streptomyces Clavuligerus
Computational-guided discovery and characterization of a sesquiterpene synthase from Streptomyces clavuligerus Jeng-Yeong Chowa,1, Bo-Xue Tianb,c,1, Gurusankar Ramamoorthya, Brandan S. Hillerichd, Ronald D. Seideld, Steven C. Almod, Matthew P. Jacobsonb,c,2, and C. Dale Poultera,2 aDepartment of Chemistry, University of Utah, Salt Lake City, UT 84112; bDepartment of Pharmaceutical Chemistry, School of Pharmacy, University of California, San Francisco, CA 94158; cCalifornia Institute for Quantitative Biomedical Research, University of California, San Francisco, CA 94158; and dDepartment of Biochemistry, Albert Einstein College of Medicine, Bronx, NY 10461 Contributed by C. Dale Poulter, March 21, 2015 (sent for review March 6, 2015) Terpenoids are a large structurally diverse group of natural products in the hydrocarbon chain. The initial cyclization is often followed with an array of functions in their hosts. The large amount of by additional cyclizations and rearrangements to ultimately pro- genomic information from recent sequencing efforts provides duce a myriad of different carbon skeletons. Class II terpene syn- opportunities and challenges for the functional assignment of thases generate an electrophilic tertiary carbocation by protonation terpene synthases that construct the carbon skeletons of these of a trisubstituted double bond mediated by an acidic residue (e.g., compounds. Inferring function from the sequence and/or structure Asp) in their active sites. These enzymes then mediate cyclization of these enzymes is not trivial -

Characterization of Natural Product Biological Imprints for Computer-Aided Drug Design Applications Noe Sturm
Characterization of natural product biological imprints for computer-aided drug design applications Noe Sturm To cite this version: Noe Sturm. Characterization of natural product biological imprints for computer-aided drug design applications. Cheminformatics. Université de Strasbourg, 2015. English. NNT : 2015STRAF059. tel-01300872 HAL Id: tel-01300872 https://tel.archives-ouvertes.fr/tel-01300872 Submitted on 11 Apr 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE STRASBOURG ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES Laboratoire d’Innovation Thérapeutique, UMR 7200 en cotutelle avec Eskitis Institute for Drug Discovery, Griffith University THÈSE présentée par Noé STURM soutenue le : 8 Décembre 2015 pour obtenir le grade de : Docteur de l’université de Strasbourg Discipline/Spécialité : Chimie/Chémoinformatique Caractérisation de l’empreinte biologique des produits naturels pour des applications de conception rationnelle de médicament assistée par ordinateur THÈSE dirigée par : KELLENBERGER Esther Professeur, Université de Strasbourg QUINN Ronald Professeur, Université de Griffith, Brisbane, Australie RAPPORTEURS : IORGA Bogdan Chargé de recherche HDR, Institut de Chimie des Substances Naturelles, Gif-sur-Yvette GÜNTHER Stefan Professeur, Université Albert Ludwigs, Fribourg, Allemagne Acknowledgments First of all, I would like to thank my two supervisors Professor Kellenberger Esther and Professor Quinn Ronald for their excellent assistance, both academic and personal in nature. -

Expression and Mechanistic Analysis of a Germacradienol Synthase from Streptomyces Coelicolor Implicated in Geosmin Biosynthesis
Expression and mechanistic analysis of a germacradienol synthase from Streptomyces coelicolor implicated in geosmin biosynthesis David E. Cane* and Rory M. Watt Department of Chemistry, Brown University, Providence, RI 02912 Communicated by Christopher T. Walsh, Harvard Medical School, Boston, MA, December 16, 2002 (received for review October 11, 2002) The PCR has been used to amplify a 2,181-bp ORF from Strepto- -SCO6073), encod ؍) myces coelicolor A3(2), designated SC9B1.20 ing a protein of 726 amino acids and showing significant sequence similarity at the deduced amino acid level in both the N-terminal and C-terminal halves to the known sesquiterpene synthase pen- talenene synthase. The full-length recombinant protein was ex- pressed at high levels in Escherichia coli and shown to catalyze the 2؉ Mg -dependent conversion of farnesyl diphosphate to the ses- Scheme 1. Cyclization of FPP 2 to pentalenene 3, catalyzed by pentalenene quiterpene alcohol (4S,7R)-germacra-1 (10)E,5E-diene-11-ol. The synthase (PS). ؊3 ؊1 enzymatic cyclization had a kcat of 6.2 ؎ 0.5 ؋ 10 s and a Km -for farnesyl diphosphate of 62 ؎ 8 nM. Expression of the N terminal (366 amino acids) domain of the SC9B1.20 protein also synthase. Indeed, microbial sesquiterpene synthases in general gave a fully functional cyclase which converted farnesyl diphos- show no overall sequence similarity either to one another (except phate to the identical sesquiterpene alcohol with a slightly lower for orthologs synthesizing the same sesquiterpene product) or to ؊3 ؊1 kcat of 3.2 ؎ 0.4 ؋ 10 s and a twofold greater km of 115 ؎ 14 any other protein. -

Introduction to Enzyme and Coenzyme Chemistry
Introduction to Enzyme and Coenzyme Chemistry SECOND EDITION T.D.H. Bugg Bugg/Introduction to Enzyme and Coenzyme Chemistry Final Proof 22.7.2004 3:52pm page i Introduction to Enzyme and Coenzyme Chemistry Bugg/Introduction to Enzyme and Coenzyme Chemistry Final Proof 22.7.2004 3:52pm page ii Bugg/Introduction to Enzyme and Coenzyme Chemistry Final Proof 22.7.2004 3:52pm page iii Introduction to Enzyme and Coenzyme Chemistry Second Edition TIM BUGG Professor of Biological Chemistry, Department of Chemistry, University of Warwick, UK Bugg/Introduction to Enzyme and Coenzyme Chemistry Final Proof 22.7.2004 3:52pm page iv ß 1997, 2004 by Blackwell Publishing Ltd Editorial oYces: Blackwell Publishing Ltd, 9600 Garsington Road, Oxford OX4 2DQ, UK Tel: þ44 (0)1865 776868 Blackwell Publishing Inc., 350 Main Street, Malden, MA 02148-5020, USA Tel: þ1 781 388 8250 Blackwell Publishing Asia Pty Ltd, 550 Swanston Street, Carlton, Victoria 3053, Australia Tel: þ61 (0)3 8359 1011 The right of the Author to be identiWed as the Author of this Work has been asserted in accordance with the Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. First published 1997 by Blackwell Science Second edition published 2004 by Blackwell Publishing Library of Congress Cataloging-in-Publication Data Bugg, Tim. -

Similar Structures to the E-To-H Helix Unit in the Globin-Like Fold Are Found in Other Helical Folds
Biomolecules 2014, 4, 268-288; doi:10.3390/biom4010268 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Similar Structures to the E-to-H Helix Unit in the Globin-Like Fold are Found in Other Helical Folds Masanari Matsuoka 1,2, Aoi Fujita 1, Yosuke Kawai 1,† and Takeshi Kikuchi 1,* 1 Department of Bioinformatics, College of Life Sciences, Ritsumeikan University, Kusatsu, Shiga 525-8577, Japan; E-Mails: [email protected] (M.M.); [email protected] (A.F.); [email protected] (Y.K.) 2 Japan Society for the Promotion of Science (JSPS), Ichibancho, Chiyoda-ku, Tokyo 102-8471, Japan † Present address: Division of Biomedical Information Analysis, Department of Integrative Genomics, Tohoku Medical Megabank Organization, Tohoku University, Sendai, Miyagi 980-8575, Japan * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-77-561-5909; Fax: +81-77-561-5203. Received: 6 December 2013; in revised form: 11 February 2014 / Accepted: 13 February 2014 / Published: 27 February 2014 Abstract: A protein in the globin-like fold contains six alpha-helices, A, B, E, F, G and H. Among them, the E-to-H helix unit (E, F, G and H helices) forms a compact structure. In this study, we searched similar structures to the E-to-H helix of leghomoglobin in the whole protein structure space using the Dali program. Several similar structures were found in other helical folds, such as KaiA/RbsU domain and Type III secretion system domain. These observations suggest that the E-to-H helix unit may be a common subunit in the whole protein 3D structure space.