Activation of Rac1 by Src-Dependent Phosphorylation of Dock180y1811 Mediates Pdgfrα-Stimulated Glioma Tumorigenesis in Mice and Humans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structure of the Dock2âˆ'elmo1 Complex Provides Insights Into
ARTICLE https://doi.org/10.1038/s41467-020-17271-9 OPEN Structure of the DOCK2−ELMO1 complex provides insights into regulation of the auto-inhibited state Leifu Chang1,7, Jing Yang1, Chang Hwa Jo 2, Andreas Boland 1,8, Ziguo Zhang 1, Stephen H. McLaughlin 1, Afnan Abu-Thuraia 3, Ryan C. Killoran2, Matthew J. Smith 2,4,9, Jean-Francois Côté 3,5,6,9 & ✉ David Barford 1 DOCK (dedicator of cytokinesis) proteins are multidomain guanine nucleotide exchange 1234567890():,; factors (GEFs) for RHO GTPases that regulate intracellular actin dynamics. DOCK proteins share catalytic (DOCKDHR2) and membrane-associated (DOCKDHR1) domains. The structurally-related DOCK1 and DOCK2 GEFs are specific for RAC, and require ELMO (engulfment and cell motility) proteins for function. The N-terminal RAS-binding domain (RBD) of ELMO (ELMORBD) interacts with RHOG to modulate DOCK1/2 activity. Here, we determine the cryo-EM structures of DOCK2−ELMO1 alone, and as a ternary complex with RAC1, together with the crystal structure of a RHOG−ELMO2RBD complex. The binary DOCK2−ELMO1 complex adopts a closed, auto-inhibited conformation. Relief of auto- inhibition to an active, open state, due to a conformational change of the ELMO1 subunit, exposes binding sites for RAC1 on DOCK2DHR2, and RHOG and BAI GPCRs on ELMO1. Our structure explains how up-stream effectors, including DOCK2 and ELMO1 phosphorylation, destabilise the auto-inhibited state to promote an active GEF. 1 MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, UK. 2 Institute for Research in Immunology and Cancer, Université de Montréal, Montréal, Québec H3T 1J4, Canada. 3 Montreal Institute of Clinical Research (IRCM), Montréal, QC H2W 1R7, Canada. -
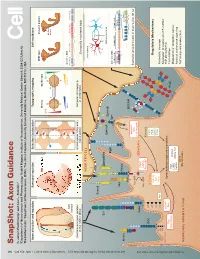
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Tyr724 Phosphorylation of ELMO1 by Src Is Involved in Cell Spreading and Migration Via Rac1 Activation
Title Tyr724 phosphorylation of ELMO1 by Src is involved in cell spreading and migration via Rac1 activation Makino, Yoshinori; Tsuda, Masumi; Ohba, Yusuke; Nishihara, Hiroshi; Sawa, Hirofumi; Nagashima, Kazuo; Tanaka, Author(s) Shinya Cell communication and signaling, 13, 35 Citation https://doi.org/10.1186/s12964-015-0113-y Issue Date 2015-07-26 Doc URL http://hdl.handle.net/2115/59828 Rights(URL) http://creativecommons.org/licenses/by/4.0 Type article File Information s12964-015-0113-y.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Makino et al. Cell Communication and Signaling (2015) 13:35 DOI 10.1186/s12964-015-0113-y RESEARCH ARTICLE Open Access Tyr724 phosphorylation of ELMO1 by Src is involved in cell spreading and migration via Rac1 activation Yoshinori Makino1,2, Masumi Tsuda1, Yusuke Ohba3, Hiroshi Nishihara4, Hirofumi Sawa1,5, Kazuo Nagashima1,6 and Shinya Tanaka1,4* Abstract Background: The complex of Dock180/ELMO1 that functions as a bipartite guanine nucleotide exchange factor for Rac is essential for diverse physiological and pathological processes of cells such as cell migration, phagocytosis, and invasion of cancer cells. Among the Src-family tyrosine kinases (SFKs), it has been reported that Hck directly phosphorylates ELMO1, regulating phagocytosis by promoting activation of Rac1; however, the involvement of other SFKs in ELMO1 phosphorylation has remained unknown. Here, we identified novel tyrosine (Y) residues of ELMO1 phosphorylated by SFKs, and examined the effects on Rac1 activity, cell adhesion, spreading, and cell motility on extracellular matrix (ECM). Results: In this study, we unveiled that Src and Fyn can induce tyrosine phosphorylation of ELMO1 in in vivo and in vitro phosphorylation assays. -

Structural Basis for Mutual Relief of the Rac Guanine Nucleotide Exchange Factor DOCK2 and Its Partner ELMO1 from Their Autoinhibited Forms
Structural basis for mutual relief of the Rac guanine nucleotide exchange factor DOCK2 and its partner ELMO1 from their autoinhibited forms Kyoko Hanawa-Suetsugua, Mutsuko Kukimoto-Niinoa,b, Chiemi Mishima-Tsumagaria, Ryogo Akasakaa, Noboru Ohsawaa, Shun-ichi Sekinea,c, Takuhiro Itoa,c, Naoya Tochioa, Seizo Koshibaa, Takanori Kigawaa,d, Takaho Teradaa,b, Mikako Shirouzua, Akihiko Nishikimib,e,f, Takehito Urunoe, Tomoya Katakaig, Tatsuo Kinashig, Daisuke Kohdah, Yoshinori Fukuib,e,f,1, and Shigeyuki Yokoyamaa,b,c,1 aRIKEN Systems and Structural Biology Center, 1-7-22 Suehiro, Tsurumi, Yokohama 230-0045, Japan; bJapan Science and Technology Agency, Core Research for Evolutional Science and Technology, Tokyo 102-0075, Japan; cDepartment of Biophysics and Biochemistry, Graduate School of Science, University of Tokyo, 7-3-1 Hongo, Bunkyo, Tokyo 113-0033, Japan; dTokyo Institute of Technology, 4259 Nagatsuta, Midori, Yokohama 226-8502, Japan; eDivision of Immunogenetics, Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, Fukuoka 812-8582, Japan; fResearch Center for Advanced Immunology, Kyushu University, Fukuoka 812-8582, Japan; gDepartment of Molecular Genetics, Institute of Biomedical Science, Kansai Medical University, Osaka 570-8506, Japan; and hDivision of Structural Biology, Medical Institute of Bioregulation, Kyushu University, Fukuoka 812-8582, Japan Edited by John Kuriyan, University of California, Berkeley, CA, and approved December 31, 2011 (received for review September 6, 2011) DOCK2, a hematopoietic cell-specific, atypical guanine nucleotide for the Rho-family GTPases. There are 11 mammalian members exchange factor, controls lymphocyte migration through ras-related (DOCK180, DOCK2-11) of the CDM family. The CDM proteins C3 botulinum toxin substrate (Rac) activation. -

Identification of Two Signaling Submodules Within the Crkii/ELMO
Cell Death and Differentiation (2007) 14, 963–972 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells A-C Tosello-Trampont1,4, JM Kinchen1,2,4, E Brugnera1,3, LB Haney1, MO Hengartner2 and KS Ravichandran*,1 Removal of apoptotic cells is a dynamic process coordinated by ligands on apoptotic cells, and receptors and other signaling proteins on the phagocyte. One of the fundamental challenges is to understand how different phagocyte proteins form specific and functional complexes to orchestrate the recognition/removal of apoptotic cells. One evolutionarily conserved pathway involves the proteins cell death abnormal (CED)-2/chicken tumor virus no. 10 (CT10) regulator of kinase (Crk)II, CED-5/180 kDa protein downstream of chicken tumor virus no. 10 (Crk) (Dock180), CED-12/engulfment and migration (ELMO) and MIG-2/RhoG, leading to activation of the small GTPase CED-10/Rac and cytoskeletal remodeling to promote corpse uptake. Although the role of ELMO : Dock180 in regulating Rac activation has been well defined, the function of CED-2/CrkII in this complex is less well understood. Here, using functional studies in cell lines, we observe that a direct interaction between CrkII and Dock180 is not required for efficient removal of apoptotic cells. Similarly, mutants of CED-5 lacking the CED-2 interaction motifs could rescue engulfment and migration defects in CED-5 deficient worms. Mutants of CrkII and Dock180 that could not biochemically interact could colocalize in membrane ruffles. -

Disassembly of the Dying: Mechanisms and Functions
Review Disassembly of the Dying: Mechanisms and Functions 1 1, Georgia K. Atkin-Smith and Ivan K.H. Poon * The disassembly of an apoptotic cell into subcellular fragments, termed Trends apoptotic bodies (ApoBDs), is a hallmark of apoptosis. Although the genera- Apoptotic cell disassembly is a highly tion of ApoBDs is generally understood as being stochastic, it is becoming complex process regulated by a series of well-coordinated morphological increasingly clear that ApoBD formation is a highly regulated process involv- steps including apoptotic membrane ing distinct morphological steps and molecular factors. Functionally, ApoBDs blebbing, apoptotic protrusion forma- fi could facilitate the ef cient clearance of apoptotic material by surrounding tion, and fragmentation. phagocytes as well as mediate the transfer of biomolecules including micro- Plasma-membrane blebbing is not the RNAs and proteins between cells to aid in intercellular communications. sole process required for apoptotic Therefore, the formation of ApoBDs is an important process downstream body (ApoBD) formation, but mem- brane protrusions including microtu- from apoptotic cell death. We discuss here the mechanisms and functions bule spikes, apoptopodia, and of apoptotic cell disassembly. beaded apoptopodia may act in con- cert to aid the generation of ApoBDs. Cell Disassembly as a Key Downstream Process of Apoptotic Cell Death The mechanism of how apoptotic cells Billions of cells undergo apoptosis (a form of programmed cell death) daily as part of disassembly can determine the quantity and quality (size and contents) of physiological homeostasis [1]. At later stages of apoptosis, some cell types can generate ApoBDs. subcellular (1–5 mm) membrane-bound extracellular vesicles termed apoptotic bodies – (ApoBDs, see Glossary) [1 3]. -

Snapshot: Axon Guidance II Alex L
SnapShot: Axon Guidance II Alex L. Kolodkin1 and R. Jeroen Pasterkamp2 1Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA 2Department of Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands Axon guidance protein Chemotrophic effect Ligand-binding receptor Receptor processing Coreceptor Receptor signaling COFILIN, LIMK1, PI3K, BMP SE Repulsion BMP-RIB, BMP-RII - - SMAD1, SMAD6 DRAXIN SE Repulsion DCC - - - a2-CHIMAERIN, EPHEXIN-1, EPHRINA GPI Repulsion EPHA ADAM10 - NCK1/2, RAC1, RHOA, SPAR, VAV2/3 NCK1, PAK, p120GAP, RHOA, EPHRINB TM Repulsion EPHB MMP2/9 - ROCK, VAV2/3 TM Repulsion EPHRINA ADAM10 P75 FYN EPHA Attraction EPHRINA - RET - CDC42, DOCK180, GRB4, NCK2, EPHB TM Repulsion EPHRINB ADAM10, PS1 - PAK, RAC1 FGF SE Attraction FGFR1 - - - CDC42, DOCK180, ENA/VASP, APP, HSPG, ERK1/2, FAK, FYN, NCK1, N- SE Attraction DCC ADAM10, PS1 ROBO1 WASP, PAK, PI3K, PIP2, PKC, NETRIN RAC1, RHOA, TRIO, TRP Attraction NEOGENIN - - - Repulsion UNC5 - (DCC) FAK, SHP2, SRC FAK, LARG, LMO4, MYOIIA, RGM GPI Repulsion NEOGENIN TACE, PS1 UNC5 p120GAP, PKC, RAS, RHOA PLEXINA TM Repulsion SEMA1a - - PEBBLE, RHO, p190RHOGAP 14-3-3e, GYC76C, MICAL, SEMA1 TM Repulsion PLEXINA - OTK NERVY, PKA SE Repulsion PLEXINB - - RAC, RHO SEMA2 Attraction PLEXINB - - - AKT, COFILIN, CDK5, CRMP, FARP, NRP1/2, CAMs, SE Repulsion PLEXINA1-4 CALPAIN1 FYN, GSK3b, LIMK1, PI3K, RAC1, RTKs, ROBO SEMA3 RAP1, RAS, RND Attraction - - NRP1/2, -

Gene Polymorphism in Development of Diabetic Kidney Disease Thoria A
Omar et al. Egyptian Journal of Medical Human Genetics (2021) 22:49 Egyptian Journal of Medical https://doi.org/10.1186/s43042-021-00167-8 Human Genetics RESEARCH Open Access Role of engulfment and cell motility 1 (ELMO1) gene polymorphism in development of diabetic kidney disease Thoria A. Omar1*, Shimaa K. Zewain2, Mohamed M. Ghonaim3, Khadija A. Refaat1 and Dalia H. Abou-Elela1 Abstract Background: Diabetic kidney disease (DKD) is a progressive kidney disease that affects diabetic patients irrespective of glycemic state or hypertension. Therefore, early detection of DKD is of critical importance. Many genome-wide association studies have identified the engulfment and cell motility 1 (ELMO1) gene as a genetic marker linked to DKD. This study aimed to investigate the association between ELMO1 rs741301 gene polymorphism and the development of DKD among Egyptian patients with type 2 diabetes mellitus (T2DM). Allele and genotype frequencies were investigated in 304 subjects by real-time PCR allelic discrimination assay: 100 DKD patients, 102 diabetic patients without DKD, and 102 healthy controls. Results: GG genotype of ELMO1 (rs741301) SNP and its allele frequencies were significantly high in all diabetic patients. GG genotype had an odds ratio (OR) of 6.095 and 95% confidence interval (CI) of 2.456–15.125, p < 0.001, while the frequent allele G had an OR of 2.366 and 95% CI of 1.450–3.859, p = 0.001. No significant difference was observed between T2DM without DKD and DKD. Conclusion: Our results could not establish an association between the ELMO1 rs741301 variant and the progression of DKD. -

Rho Guanine Nucleotide Exchange Factors: Regulators of Rho Gtpase Activity in Development and Disease
Oncogene (2014) 33, 4021–4035 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease DR Cook1, KL Rossman2,3 and CJ Der1,2,3 The aberrant activity of Ras homologous (Rho) family small GTPases (20 human members) has been implicated in cancer and other human diseases. However, in contrast to the direct mutational activation of Ras found in cancer and developmental disorders, Rho GTPases are activated most commonly in disease by indirect mechanisms. One prevalent mechanism involves aberrant Rho activation via the deregulated expression and/or activity of Rho family guanine nucleotide exchange factors (RhoGEFs). RhoGEFs promote formation of the active GTP-bound state of Rho GTPases. The largest family of RhoGEFs is comprised of the Dbl family RhoGEFs with 70 human members. The multitude of RhoGEFs that activate a single Rho GTPase reflects the very specific role of each RhoGEF in controlling distinct signaling mechanisms involved in Rho activation. In this review, we summarize the role of Dbl RhoGEFs in development and disease, with a focus on Ect2 (epithelial cell transforming squence 2), Tiam1 (T-cell lymphoma invasion and metastasis 1), Vav and P-Rex1/2 (PtdIns(3,4,5)P3 (phosphatidylinositol (3,4,5)-triphosphate)-dependent Rac exchanger). Oncogene (2014) 33, 4021–4035; doi:10.1038/onc.2013.362; published online 16 September 2013 Keywords: Rac1; RhoA; Cdc42; guanine nucleotide exchange factors; cancer; -

ELMO1 Signaling Is a Promoter of Osteoclast Function and Bone Loss ✉ Sanja Arandjelovic 1 , Justin S
ARTICLE https://doi.org/10.1038/s41467-021-25239-6 OPEN ELMO1 signaling is a promoter of osteoclast function and bone loss ✉ Sanja Arandjelovic 1 , Justin S. A. Perry 1, Ming Zhou 2, Adam Ceroi3, Igor Smirnov4, Scott F. Walk1, Laura S. Shankman1, Isabelle Cambré3, Suna Onengut-Gumuscu 5, Dirk Elewaut 3, Thomas P. Conrads 2 & ✉ Kodi S. Ravichandran 1,3 Osteoporosis affects millions worldwide and is often caused by osteoclast induced bone loss. 1234567890():,; Here, we identify the cytoplasmic protein ELMO1 as an important ‘signaling node’ in osteoclasts. We note that ELMO1 SNPs associate with bone abnormalities in humans, and that ELMO1 deletion in mice reduces bone loss in four in vivo models: osteoprotegerin deficiency, ovariectomy, and two types of inflammatory arthritis. Our transcriptomic analyses coupled with CRISPR/Cas9 genetic deletion identify Elmo1 associated regulators of osteoclast function, including cathepsin G and myeloperoxidase. Further, we define the ‘ELMO1 inter- actome’ in osteoclasts via proteomics and reveal proteins required for bone degradation. ELMO1 also contributes to osteoclast sealing zone on bone-like surfaces and distribution of osteoclast-specific proteases. Finally, a 3D structure-based ELMO1 inhibitory peptide reduces bone resorption in wild type osteoclasts. Collectively, we identify ELMO1 as a signaling hub that regulates osteoclast function and bone loss, with relevance to osteoporosis and arthritis. 1 Center for Cell Clearance, Department of Microbiology, Immunology, and Cancer Biology and Carter Immunology Center, University of Virginia, Charlottesville, VA, USA. 2 Inova Schar Cancer Institute, Inova Center for Personalized Health, Fairfax, VA, USA. 3 Inflammation Research Centre, VIB, and the Department of Biomedical Molecular Biology, Ghent Univeristy, Ghent, Belgium.