Ogf-Ogfr Axis and Its Inhibitory Actions on Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Tranexamic Acid Inhibits the Plasma and Non-Irradiated Skin Markers Of
Biomedicine & Pharmacotherapy 107 (2018) 54–58 Contents lists available at ScienceDirect Biomedicine & Pharmacotherapy journal homepage: www.elsevier.com/locate/biopha Original Article Tranexamic acid inhibits the plasma and non-irradiated skin markers of photoaging induced by long-term UVA eye irradiation in female mice T ⁎ Keiichi Hiramotoa, , Yurika Yamatea, Daijiro Sugiyamab, Kazunari Matsudab, Yasutaka Iizukab, Tomohiko Yamaguchib a Department of Pharmaceutical Sciences, Suzuka University of Medical Science, 3500-3 Minamitamagakicho, Suzuka, Mie, 513-8670, Japan b R&D Department, Daiichi Sankyo Healthcare Co., LTD., 3-14-10 Nihonbashi, Chuo-ku, Tokyo, 103-8234, Japan ARTICLE INFO ABSTRACT Keywords: Photoaging can be induced by long-term ultraviolet (UV)A eye irradiation, but an ameliorating method for such Tranexamic acid photoaging is not known. In this study, we examined the effects of tranexamic acid (trans-4-aminomethylcy- Photoaging clohexanecarboxylic acid) on photoaging of the skin induced by UVA eye irradiation. We used the C57BL/6 j Urocortin 2 female mice and locally exposed their eyes to UVA at a dose of 110 kJ/m2 using an FL20SBLB-A lamp multiple β-Endorphin times a week for one year. The plasma urocortin 2, β-endorphin, methionine enkephalin (OGF), and histamine Methionine encephalin content, as well as the expression of the corticotropin-releasing hormone receptor (CRHR) type 2, μ-opioid Histamine Estrogen receptor-β receptor, opioid growth factor receptor (OGFR), T-bet, and GATA3 increased in the mice subjected to UVA eye irradiation. However, the increased levels of urocortin 2, methionine enkephalin, histamine, OGFR, T-bet, and GATA3 were suppressed by tranexamic acid treatment. -

G Protein-Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. British Journal of Pharmacology (2015) 172, 5744–5869 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: G protein-coupled receptors Stephen PH Alexander1, Anthony P Davenport2, Eamonn Kelly3, Neil Marrion3, John A Peters4, Helen E Benson5, Elena Faccenda5, Adam J Pawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, 2Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/doi/ 10.1111/bph.13348/full. G protein-coupled receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage-gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. -

G Protein‐Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology (2019) 176, S21–S141 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors Stephen PH Alexander1 , Arthur Christopoulos2 , Anthony P Davenport3 , Eamonn Kelly4, Alistair Mathie5 , John A Peters6 , Emma L Veale5 ,JaneFArmstrong7 , Elena Faccenda7 ,SimonDHarding7 ,AdamJPawson7 , Joanna L Sharman7 , Christopher Southan7 , Jamie A Davies7 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University, Parkville, Victoria 3052, Australia 3Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK 4School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 5Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 6Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 7Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Interaction of Opioid Growth Factor (OGF) and Opioid Antagonist And
International Immunopharmacology 75 (2019) 105785 Contents lists available at ScienceDirect International Immunopharmacology journal homepage: www.elsevier.com/locate/intimp Review Interaction of opioid growth factor (OGF) and opioid antagonist and their significance in cancer therapy T ⁎ ⁎ Ruizhe Wanga, Yi Zhanga, , Fengping Shanb, a Department of Gynecology, No.1 Teaching Hospital, China Medical University, Shenyang 110001, China b Department of immunology, School of Basic Medical Science, China Medical University, Shenyang, 110122, China ARTICLE INFO ABSTRACT Keywords: Endogenous opioids are neuro-peptides with multifunctional properties. Historically, opioids are used to mediate Opioid growth factor (OGF) pain; however, excess opiate consumption can lead to addiction. One endogenous opioid, methionine enkephalin Opioid antagonist (MENK), was reported to modulate cell growth, MENK was identified as an opioid growth factor (OGF) that Methionine enkephalin(MENK) interacts with the OGF receptor (OGFr) and regulates cell proliferation. Further, opioid antagonists, including Low dose naltrexone (LDN) naltrexone and naloxone are widely used to reverse drug and alcohol overdoses. Naltrexone (NTX) acts on all Cancer therapy opioid receptors, blocking the interaction between OGF and OGFr, and thus influencing cell growth. During the Immuno-regulation last decades, insights have been made concerning the interaction between OGF and OGFr, confirming that both opioids and opioid antagonists have an important role in balancing host homeostasis, host immunity and mediating cancer therapy. This review provides insight into the interactions between OGF and OGFr in the treatment of cancers. 1. Introduction their binding to the opiate receptors [5,14]. In the treatment of ad- diction, enkephalins bind with the mu or delta receptors inhibiting the 1.1. -

Blockade of the Ogf-Ogfr Axis Enhances Repair Processes In
The Pennsylvania State University The Graduate School Department of Neural and Behavioral Sciences BLOCKADE OF THE OGF-OGFR AXIS ENHANCES REPAIR PROCESSES IN DIABETIC SKIN AND BONE A Dissertation in Anatomy by Michelle B. Titunick ©2018 Michelle B. Titunick Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2018 The dissertation of Michelle B. Titunick was reviewed and approved* by the following: Patricia J. McLaughlin Professor of Neural and Behavioral Sciences Director of the Graduate Program in Anatomy Dissertation Advisor Chair of Committee Ian S. Zagon Distinguished Professor of Neural and Behavioral Sciences Gregory S. Lewis Assistant Professor of Orthopaedics and Rehabilitation Christopher Niyibizi Associate Professor of Orthopaedics and Rehabilitation *Signatures are on file in the Graduate School. ii ABSTRACT Almost 9.5% of the U.S. population had diabetes in 2015 with 5-10% having type 1 diabetes. In addition to diabetic complications such as retinopathy, nephropathy, neuropathy, and cardiovascular disease, there is also poor wound healing and delayed fracture healing. Preclinical studies from our laboratory have demonstrated that topical naltrexone (NTX) is an effective and safe treatment for diabetic complications such as corneal keratopathy, dry eye, and non-healing ulcers. Naltrexone is an opioid receptor antagonist that, at an appropriate dosage, blocks the opioid growth factor (OGF)-opioid growth factor receptor (OGFr) axis resulting in increased cell proliferation. [Met5]- enkephalin, or OGF, is a highly conserved endogenous opioid peptide that tonically regulates cell proliferation by delaying the G0/G1 phase of the cell cycle. Both systemic and topical NTX have been shown to enhance corneal epithelial wound healing in normal and diabetic rats, diabetic rabbits, as well as the type 2 diabetic model, db/db. -

Selective Opioid Growth Factor Receptor Antagonists Based on a Stilbene Isostere
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Bioorg Med Manuscript Author Chem. Author Manuscript Author manuscript; available in PMC 2018 August 15. Published in final edited form as: Bioorg Med Chem. 2017 August 15; 25(16): 4464–4474. doi:10.1016/j.bmc.2017.06.035. Selective opioid growth factor receptor antagonists based on a stilbene isostere David P. Stockdalea, Michelle B. Titunickb, Jessica M. Bieglerc,d, Jessie L. Reedc,d, Alyssa M. Hartunga, David F. Wiemera, Patricia J. McLaughlinb,d, and Jeffrey D. Neighborsc,d aDepartment of Chemistry, The University of Iowa, Iowa City, IA 52242-1294, United States bDepartment of Neural and Behavioral Sciences, Pennsylvania State University College of Medicine, Hershey, PA 17033, United States cDepartment of Pharmacology and Medicine, Pennsylvania State University College of Medicine, Hershey, PA 17033, United States dPennsylvania State University Cancer Institute, Hershey, PA 17033, United States Abstract As part of an ongoing drug development effort aimed at selective opioid receptor ligands based on the pawhuskin natural products we have synthesized a small set of amide isosteres. These amides were centered on lead compounds which are selective antagonists for the delta and kappa opioid receptors. The amide isomers revealed here show dramatically different activity from the parent stilbene compounds. Three of the isomers synthesized showed antagonist activity for the opioid growth factor (OGF)/opioid growth factor receptor (OGFR) axis which is involved in cellular and organ growth control. This cellular signaling mechanism is targeted by “low-dose” naltrexone therapy which is being tested clinically for multiple sclerosis, Crohn’s disease, cancer, and wound healing disorders. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -
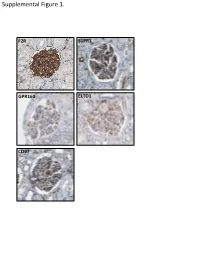
Supplemental Data
Supplemental Figure 1. F2R S1PR1 GPR160 ELTD1 CD97 Supplemental Figure 2. A B brain heart lung liver kidney spleen testis skm Expression in mouse ssues glom rok Gprc5a Gapdh Human Protein Atlas RNAseq database Supplemental Figure 3. A Gprc5a Pdgfrb Merged CL CL Gprc5a CD31 Merged CL CL B C 1.4 * 1.2 mc 1 matrix 0.8 0.6 0.4 matrix 0.2 0 pod end 1 2 mes3 D Vector Gprc5a E 40 kD - - Gprc5a - acn Supplemental Figure 4. Control 12 month-old KO 12 month-old A B C D E F 1 2 0.8 * 1.5 score 0.6 m µ 1 0.4 0.2 Slits/ 0.5 Mesangial 0 0 Ctrl KO Ctrl KO Supplemental Figure 5. A B 1.2 Vector Gprc5a 1 * 0.8 0.6 * 0.4 * Normalized Density 0.2 0 pEGFR/tEGR pSmad/tSmad TGF-β1 C 1.8 siCON 1.6 siGprc5a * 1.4 * * 1.2 1 0.8 0.6 NormalizedDensity 0.4 0.2 0 pEGFR/tEGR pSmad/tSmad TGF-β1 Supplemental table 1. List of glomerulus-expressed GPCRs as detected by qPCR. Data shown as mean ± standard deviation (Glom=glomerulus, Rok=rest of kidney). GPCR Glom Rok Glom/Rok LPAR6 41892,11 ± 38478,89 1040,06 ± 1370,12 39,28 ELTD1 30275,64 ± 14085,26 33,23 ± 46,99 910,13 GPR116 24020,06 ± 7789,84 55,1 ± 47,75 434,96 PTH1R 15402,81 ± 17644,32 7521,01 ± 3264,57 1,05 CALCRL 14096,09 ± 3854,84 199,06 ± 222,52 69,81 HPRT1 13342,04 ± 10677,69 1824,77 ± 1767,23 6,31 S1PR5 9474,7 ± 10124,3 110,84 ± 29,54 84,48 LPHN2 8645,89 ± 914,74 256,04 ± 293,87 32,77 FZD1 8176,2 ± 4947,45 1321,97 ± 1311,91 5,18 CXCR4 7097,31 ± 4388,91 535,98 ± 640,28 12,24 GPR160 6446,59 ± 1550,07 4816,3 ± 5918,99 0,34 NPY1R 6177,74 ± 6282,76 209,64 ± 296,48 28,47 PTGER4 5323,66 ± 3789,51 179,13 ± 210 28,72 RXFP1 -

Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C)
G C A T T A C G G C A T genes Article Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C) Aodán Laighneach 1 , Lieve Desbonnet 2, John P. Kelly 2, Gary Donohoe 1 and Derek W. Morris 1,* 1 Centre for Neuroimaging, Cognition and Genomics, Discipline of Biochemistry and School of Psychology, National University of Ireland Galway, H91 TK33 Galway, Ireland; [email protected] (A.L.); [email protected] (G.D.) 2 Discipline of Pharmacology and Therapeutics, National University of Ireland Galway, H91 TK33 Galway, Ireland; [email protected] (L.D.); [email protected] (J.P.K.) * Correspondence: [email protected] Abstract: Maternal immune activation (MIA) is a known risk factor for schizophrenia (SCZ) and autism spectrum disorder (ASD) and is often modelled in animal studies in order to study the effect of prenatal infection on brain function including behaviour and gene expression. Although the effect of MIA on gene expression are highly heterogeneous, combining data from multiple gene expression studies in a robust method may shed light on the true underlying biological effects caused by MIA and this could inform studies of SCZ and ASD. This study combined four RNA-seq and microarray datasets in an overlap analysis and ranked meta-analysis in order to investigate genes, pathways and cell types dysregulated in the MIA mouse models. Genes linked to SCZ and ASD and crucial in neurodevelopmental processes including neural tube folding, regulation of cellular stress and neuronal/glial cell differentiation were among the most consistently dysregulated in these ranked analyses. -

G Protein‐Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology (2019) 176, S21–S141 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors Stephen PH Alexander1 , Arthur Christopoulos2 , Anthony P Davenport3 , Eamonn Kelly4, Alistair Mathie5 , John A Peters6 , Emma L Veale5 ,JaneFArmstrong7 , Elena Faccenda7 ,SimonDHarding7 ,AdamJPawson7 , Joanna L Sharman7 , Christopher Southan7 , Jamie A Davies7 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University, Parkville, Victoria 3052, Australia 3Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK 4School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 5Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 6Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 7Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website.