Supplementary Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Loss of At-Catenin Alters the Hybrid Adhering Junctions in the Heart and Leads to Dilated Cardiomyopathy and Ventricular Arrhythmia Following Acute Ischemia
1058 Research Article Loss of aT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia Jifen Li1,*, Steven Goossens2,3,`, Jolanda van Hengel2,3,`, Erhe Gao1, Lan Cheng1, Koen Tyberghein2,3, Xiying Shang1, Riet De Rycke2, Frans van Roy2,3,* and Glenn L. Radice1 1Center for Translational Medicine, Department of Medicine, Thomas Jefferson University, Philadelphia, PA, USA 2Department for Molecular Biomedical Research, Flanders Institute for Biotechnology (VIB), B-9052 Ghent, Belgium 3Department of Biomedical Molecular Biology, Ghent University, B-9052 Ghent, Belgium *Authors for correspondence ([email protected]; [email protected]) `These authors contributed equally to this work Accepted 4 October 2011 Journal of Cell Science 125, 1058–1067 ß 2012. Published by The Company of Biologists Ltd doi: 10.1242/jcs.098640 Summary It is generally accepted that the intercalated disc (ICD) required for mechano-electrical coupling in the heart consists of three distinct junctional complexes: adherens junctions, desmosomes and gap junctions. However, recent morphological and molecular data indicate a mixing of adherens junctional and desmosomal components, resulting in a ‘hybrid adhering junction’ or ‘area composita’. The a-catenin family member aT-catenin, part of the N-cadherin–catenin adhesion complex in the heart, is the only a-catenin that interacts with the desmosomal protein plakophilin-2 (PKP2). Thus, it has been postulated that aT-catenin might serve as a molecular integrator of the two adhesion complexes in the area composita. To investigate the role of aT-catenin in the heart, gene targeting technology was used to delete the Ctnna3 gene, encoding aT-catenin, in the mouse. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -
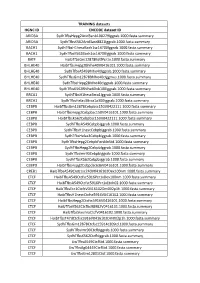
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

List of Genes Used in Cell Type Enrichment Analysis
List of genes used in cell type enrichment analysis Metagene Cell type Immunity ADAM28 Activated B cell Adaptive CD180 Activated B cell Adaptive CD79B Activated B cell Adaptive BLK Activated B cell Adaptive CD19 Activated B cell Adaptive MS4A1 Activated B cell Adaptive TNFRSF17 Activated B cell Adaptive IGHM Activated B cell Adaptive GNG7 Activated B cell Adaptive MICAL3 Activated B cell Adaptive SPIB Activated B cell Adaptive HLA-DOB Activated B cell Adaptive IGKC Activated B cell Adaptive PNOC Activated B cell Adaptive FCRL2 Activated B cell Adaptive BACH2 Activated B cell Adaptive CR2 Activated B cell Adaptive TCL1A Activated B cell Adaptive AKNA Activated B cell Adaptive ARHGAP25 Activated B cell Adaptive CCL21 Activated B cell Adaptive CD27 Activated B cell Adaptive CD38 Activated B cell Adaptive CLEC17A Activated B cell Adaptive CLEC9A Activated B cell Adaptive CLECL1 Activated B cell Adaptive AIM2 Activated CD4 T cell Adaptive BIRC3 Activated CD4 T cell Adaptive BRIP1 Activated CD4 T cell Adaptive CCL20 Activated CD4 T cell Adaptive CCL4 Activated CD4 T cell Adaptive CCL5 Activated CD4 T cell Adaptive CCNB1 Activated CD4 T cell Adaptive CCR7 Activated CD4 T cell Adaptive DUSP2 Activated CD4 T cell Adaptive ESCO2 Activated CD4 T cell Adaptive ETS1 Activated CD4 T cell Adaptive EXO1 Activated CD4 T cell Adaptive EXOC6 Activated CD4 T cell Adaptive IARS Activated CD4 T cell Adaptive ITK Activated CD4 T cell Adaptive KIF11 Activated CD4 T cell Adaptive KNTC1 Activated CD4 T cell Adaptive NUF2 Activated CD4 T cell Adaptive PRC1 Activated -

The Classification of Esterases: an Important Gene Family Involved in Insecticide Resistance - a Review
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 107(4): 437-449, June 2012 437 The classification of esterases: an important gene family involved in insecticide resistance - A Review Isabela Reis Montella1,2, Renata Schama1,2,3/+, Denise Valle1,2,3 1Laboratório de Fisiologia e Controle de Artrópodes Vetores, Instituto Oswaldo Cruz-Fiocruz, Av. Brasil 4365, 21040-900 Rio de Janeiro, RJ, Brasil 2Instituto de Biologia do Exército, Rio de Janeiro, RJ, Brasil 3Instituto Nacional de Ciência e Tecnologia em Entomologia Molecular, Rio de Janeiro, RJ, Brasil The use of chemical insecticides continues to play a major role in the control of disease vector populations, which is leading to the global dissemination of insecticide resistance. A greater capacity to detoxify insecticides, due to an increase in the expression or activity of three major enzyme families, also known as metabolic resistance, is one major resistance mechanisms. The esterase family of enzymes hydrolyse ester bonds, which are present in a wide range of insecticides; therefore, these enzymes may be involved in resistance to the main chemicals employed in control programs. Historically, insecticide resistance has driven research on insect esterases and schemes for their classification. Currently, several different nomenclatures are used to describe the esterases of distinct species and a universal standard classification does not exist. The esterase gene family appears to be rapidly evolving and each insect species has a unique complement of detoxification genes with only a few orthologues across species. The examples listed in this review cover different aspects of their biochemical nature. However, they do not appear to contribute to reliably distinguish among the different resistance mechanisms. -

Genome-Wide Identification and Analysis of Prognostic Features in Human Cancers
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446243; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Genome-wide identification and analysis of prognostic features in human cancers Joan C. Smith1,2 and Jason M. Sheltzer1* 1. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724. 2. Google, Inc., New York, NY 10011. * Lead contact; to whom correspondence may be addressed. E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446243; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Clinical decisions in cancer rely on precisely assessing patient risk. To improve our ability to accurately identify the most aggressive malignancies, we constructed genome-wide survival models using gene expression, copy number, methylation, and mutation data from 10,884 patients with known clinical outcomes. We identified more than 100,000 significant prognostic biomarkers and demonstrate that these genomic features can predict patient outcomes in clinically-ambiguous situations. While adverse biomarkers are commonly believed to represent cancer driver genes and promising therapeutic targets, we show that cancer features associated with shorter survival times are not enriched for either oncogenes or for successful drug targets. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 As
cells Article Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners 1, 1, 2, Eunate Gallardo-Vara y, Lidia Ruiz-Llorente y, Juan Casado-Vela y , 3 4 5 6, , María J. Ruiz-Rodríguez , Natalia López-Andrés , Asit K. Pattnaik , Miguel Quintanilla z * 1, , and Carmelo Bernabeu z * 1 Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas (CSIC), and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), 28040 Madrid, Spain; [email protected] (E.G.-V.); [email protected] (L.R.-L.) 2 Bioengineering and Aerospace Engineering Department, Universidad Carlos III and Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED), Leganés, 28911 Madrid, Spain; [email protected] 3 Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain; [email protected] 4 Cardiovascular Translational Research, Navarrabiomed, Complejo Hospitalario de Navarra (CHN), Universidad Pública de Navarra (UPNA), IdiSNA, 31008 Pamplona, Spain; [email protected] 5 School of Veterinary Medicine and Biomedical Sciences, and Nebraska Center for Virology, University of Nebraska-Lincoln, Lincoln, NE 68583, USA; [email protected] 6 Instituto de Investigaciones Biomédicas “Alberto Sols”, Consejo Superior de Investigaciones Científicas (CSIC), and Departamento de Bioquímica, Universidad Autónoma de Madrid (UAM), 28029 Madrid, Spain * Correspondence: [email protected] (M.Q.); [email protected] (C.B.) These authors contributed equally to this work. y Equal senior contribution. z Received: 7 August 2019; Accepted: 7 September 2019; Published: 13 September 2019 Abstract: Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Conditional Ablation of P130cas/BCAR1 Adaptor Protein
Camacho Leal et al. Cell Communication and Signaling (2018) 16:73 https://doi.org/10.1186/s12964-018-0289-z RESEARCH Open Access Conditional ablation of p130Cas/BCAR1 adaptor protein impairs epidermal homeostasis by altering cell adhesion and differentiation Maria del Pilar Camacho Leal†, Andrea Costamagna†, Beatrice Tassone, Stefania Saoncella, Matilde Simoni, Dora Natalini, Aurora Dadone, Marianna Sciortino, Emilia Turco, Paola Defilippi, Enzo Calautti‡ and Sara Cabodi*‡ Abstract Background: p130 Crk-associated substrate (p130CAS; also known as BCAR1) is a scaffold protein that modulates many essential cellular processes such as cell adhesion, proliferation, survival, cell migration, and intracellular signaling. p130Cas has been shown to be highly expressed in a variety of human cancers of epithelial origin. However, few data are available regarding the role of p130Cas during normal epithelial development and homeostasis. Methods: To this end, we have generated a genetically modified mouse in which p130Cas protein was specifically ablated in the epidermal tissue. Results: By using this murine model, we show that p130Cas loss results in increased cell proliferation and reduction of cell adhesion to extracellular matrix. In addition, epidermal deletion of p130Cas protein leads to premature expression of “late” epidermal differentiation markers, altered membrane E-cadherin/catenin proteins localization and aberrant tyrosine phosphorylation of E-cadherin/catenin complexes. Interestingly, these alterations in adhesive properties in absence