Deciphering the Wisent Demographic and Adaptive Histories From
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Olfactory Receptors in Non-Chemosensory Tissues
BMB Reports Invited Mini Review Olfactory receptors in non-chemosensory tissues NaNa Kang & JaeHyung Koo* Department of Brain Science, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 711-873, Korea Olfactory receptors (ORs) detect volatile chemicals that lead to freezing behavior (3-5). the initial perception of smell in the brain. The olfactory re- ORs are localized in the cilia of olfactory sensory neurons ceptor (OR) is the first protein that recognizes odorants in the (OSNs) in the olfactory epithelium (OE) and are activated by olfactory signal pathway and it is present in over 1,000 genes chemical cues, typically odorants at the molecular level, in mice. It is also the largest member of the G protein-coupled which lead to the perception of smell in the brain (6). receptors (GPCRs). Most ORs are extensively expressed in the Tremendous research was conducted since Buck and Axel iso- nasal olfactory epithelium where they perform the appropriate lated ORs as an OE-specific expression in 1991 (7). OR genes, physiological functions that fit their location. However, recent the largest family among the G protein-coupled receptors whole-genome sequencing shows that ORs have been found (GPCRs) (8), constitute more than 1,000 genes on the mouse outside of the olfactory system, suggesting that ORs may play chromosome (9, 10) and more than 450 genes in the human an important role in the ectopic expression of non-chemo- genome (11, 12). sensory tissues. The ectopic expressions of ORs and their phys- Odorant activation shows a distinct signal transduction iological functions have attracted more attention recently since pathway for odorant perception. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

2U12/U37116 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 22 March 2012 (22.03.2012) 2U12/U37116 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12Q 1/68 (2006.01) A61K 38/20 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US201 1/05 1385 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 13 September 201 1 (13.09.201 1) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, (26) Publication Language: English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (30) Priority Data: ZM, ZW. 61/382,231 13 September 2010 (13.09.2010) US (84) Designated States (unless otherwise indicated, for every (71) Applicant (for all designated States except US): THE kind of regional protection available): ARIPO (BW, GH, CHILDREN'S HOSPITAL OF PHILADELPHIA [US/ GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, US]; Research Institute, Abramson Research Center, Suite ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, 140, Office of Technology Transfer, 3615 Civic Center TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Boulevard, Philadelphia, PA 19104 (US). -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Download from the Sequence Read Archive Repository ( 421 Under the Accession Number SRP070526
bioRxiv preprint doi: https://doi.org/10.1101/058446; this version posted June 12, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Article 2 Discoveries 3 4 Title 5 Deciphering the wisent demographic and adaptive histories from individual 6 whole-genome sequences 7 8 Authors and Affiliations 9 Mathieu GAUTIER1,2§, Katayoun MOAZAMI-GOUDARZI3, Hubert LEVEZIEL4, Hugues 10 PARINELLO5, Cécile GROHS3, Stéphanie RIALLE5, Rafał KOWALCZYK6, Laurence 11 FLORI3,7§ 12 1 CBGP, INRA, CIRAD, IRD, Supagro, 34988 Montferrier-sur-Lez, France 13 2 IBC, Institut de Biologie Computationnelle, 34095 Montpellier, France 14 3 GABI, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France 15 4 UGMA, INRA, Université de Limoges, 87060 Limoges, France 16 5 MGX-Montpellier GenomiX, c/o Institut de Génomique Fonctionnelle, Montpellier, France 17 6 Mammal Research Institute, Polish Academy of Sciences, 17-230 Białowieża, Poland 18 7 INTERTRYP, CIRAD, IRD, 34398 Montpellier, France 19 20 §Corresponding authors 21 Emails: 22 [email protected] 23 [email protected] 24 1 bioRxiv preprint doi: https://doi.org/10.1101/058446; this version posted June 12, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 25 Abstract 26 As the largest European herbivore, the wisent (Bison bonasus) is emblematic of the continent 27 wildlife but has unclear origins. Here, we infer its demographic and adaptive histories from 28 two individual whole genome sequences via a detailed comparative analysis with bovine 29 genomes. -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

OR1M1 Sirna (H): Sc-97898
SANTA CRUZ BIOTECHNOLOGY, INC. OR1M1 siRNA (h): sc-97898 BACKGROUND CHROMOSOMAL LOCATION Olfactory receptors interact with odorant molecules in the nose to initiate Genetic locus: OR1M1 (human) mapping to 19p13.2. a neuronal response that leads to the perception of smell. While they share a seven transmembrane domain structure with many neurotransmitter and PRODUCT hormone receptors, olfactory receptors are responsible for the recognition OR1M1 siRNA (h) is a pool of 3 target-specific 19-25 nt siRNAs designed to and transduction of odorant signals. The olfactory receptor gene family is knock down gene expression. Each vial contains 3.3 nmol of lyophilized the largest in the genome. OR1M1 (olfactory receptor 1M1), also known as siRNA, sufficient for a 10 µM solution once resuspended using protocol OR19-5 or OR19-6, is a 313 amino acid multi-pass membrane protein that below. Suitable for 50-100 transfections. Also see OR1M1 shRNA Plasmid (h): belongs to the G protein-coupled receptor 1 family. The gene that encodes sc-97898-SH and OR1M1 shRNA (h) Lentiviral Particles: sc-97898-V as alter- OR1M1 consists of nearly 1,000 bases and maps to human chromosome nate gene silencing products. 19p13.2. Chromosome 19 consists of over 63 million bases, houses approxi- mately 1,400 genes and is recognized for having the greatest gene density For independent verification of OR1M1 (h) gene silencing results, we also of the human chromosomes. It is the genetic home for a number of immu- provide the individual siRNA duplex components. Each is available as noglobulin (Ig) superfamily members, including the killer cell and leukocyte 3.3 nmol of lyophilized siRNA. -
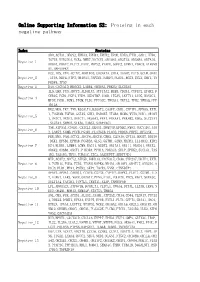
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Identifying Drug Sensitivity Subnetworks with NETPHIX
bioRxiv preprint doi: https://doi.org/10.1101/543876. this version posted February 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. It is made available under a CC-BY-NC-ND 4.0 International license. Identifying Drug Sensitivity Subnetworks with NETPHLIX Yoo-Ah Kim *† Rebecca Sarto Basso *†‡ Damian Wojtowicz † Dorit S. Hochbaum ‡ Fabio Vandin §¶ Teresa M. Prztycka †¶ Abstract Phenotypic heterogeneity in cancer is often caused by different patterns of genetic alterations. Under- standing such phenotype-genotype relationships is fundamental for the advance of personalized medicine. One of the important challenges in the area is to predict drug response on a personalized level. The pathway- centric view of cancer significantly advanced the understanding of genotype-phenotype relationships. How- ever, most of network identification methods in cancer focus on identifying subnetworks that include gen- eral cancer drivers or are associated with discrete features such as cancer subtypes, hence cannot be applied directly for the analysis of continuous features like drug response. On the other hand, existing genome wide association approaches do not fully utilize the complex proprieties of cancer mutational landscape. To ad- dress these challenges, we propose a computational method, named NETPHLIX (NETwork-to-PHenotpe mapping LeveragIng eXlusivity), which aims to identify mutated subnetworks that are associated with drug response (or any continuous cancer phenotype). Utilizing properties such as mutual exclusivity and interac- tions among genes, we formulate the problem as an integer linear program and solve it optimally to obtain a set of genes satisfying the constraints. NETPHLIX identified gene modules significantly associated with many drugs, including interesting response modules to MEK1/2 inhibitors in both directions (increased and decreased sensitivity to the drug) that the previous method, which does not utilize network informa- tion, failed to identify. -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI.