Regulated Control of Gene Therapies with a Drug Induced Switch
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SF3B3) and Sin3a Associated Protein 130 (SAP130
cells Communication Ambiguity about Splicing Factor 3b Subunit 3 (SF3B3) and Sin3A Associated Protein 130 (SAP130) Paula I. Metselaar 1,* , Celine Hos 1, Olaf Welting 1, Jos A. Bosch 2,3, Aletta D. Kraneveld 4 , Wouter J. de Jonge 1 and Anje A. Te Velde 1 1 Tytgat Institute for Liver and Intestinal Research, AGEM, Amsterdam UMC, University of Amsterdam, 1105BK Amsterdam, The Netherlands; [email protected] (C.H.); [email protected] (O.W.); [email protected] (W.J.d.J.); [email protected] (A.A.T.V.) 2 Department of Psychology, University of Amsterdam, 1018WS Amsterdam, The Netherlands; [email protected] 3 Department of Medical Psychology, Amsterdam UMC, University of Amsterdam, 1001NK Amsterdam, The Netherlands 4 Division of Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University, 3584CG Utrecht, The Netherlands; [email protected] * Correspondence: [email protected] Abstract: In 2020, three articles were published on a protein that can activate the immune system by binding to macrophage-inducible C-type lectin receptor (Mincle). In the articles, the protein was referred to as ‘SAP130, a subunit of the histone deacetylase complex.’ However, the Mincle ligand the authors aimed to investigate is splicing factor 3b subunit 3 (SF3B3). This splicing factor is unrelated to SAP130 (Sin3A associated protein 130, a subunit of the histone deacetylase-dependent Sin3A corepressor complex). The conclusions in the three articles were formulated for SF3B3, Citation: Metselaar, P.I.; Hos, C.; while the researchers used qPCR primers and antibodies against SAP130. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features
cancers Review Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features Valeria Visconte 1, Megan O. Nakashima 2 and Heesun J. Rogers 2,* 1 Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] 2 Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-445-2719 Received: 15 October 2019; Accepted: 19 November 2019; Published: 22 November 2019 Abstract: Components of the pre-messenger RNA splicing machinery are frequently mutated in myeloid malignancies. Mutations in LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2 genes occur at various frequencies ranging between 40% and 85% in different subtypes of myelodysplastic syndrome (MDS) and 5% and 10% of acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs). In some instances, splicing factor (SF) mutations have provided diagnostic utility and information on clinical outcomes as exemplified by SF3B1 mutations associated with increased ring sideroblasts (RS) in MDS-RS or MDS/MPN-RS with thrombocytosis. SF3B1 mutations are associated with better survival outcomes, while SRSF2 mutations are associated with a shorter survival time and increased AML progression, and U2AF1 mutations with a lower remission rate and shorter survival time. Beside the presence of mutations, transcriptomics technologies have shown that one third of genes in AML patients are differentially expressed, leading to altered transcript stability, interruption of protein function, and improper translation compared to those of healthy individuals. The detection of SF mutations demonstrates the importance of splicing abnormalities in the hematopoiesis of MDS and AML patients given the fact that abnormal splicing regulates the function of several transcriptional factors (PU.1, RUNX1, etc.) crucial in hematopoietic function. -
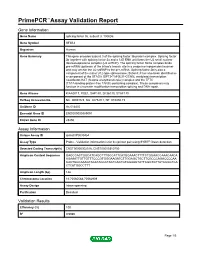
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name splicing factor 3b, subunit 3, 130kDa Gene Symbol SF3B3 Organism Human Gene Summary This gene encodes subunit 3 of the splicing factor 3b protein complex. Splicing factor 3b together with splicing factor 3a and a 12S RNA unit forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP). The splicing factor 3b/3a complex binds pre-mRNA upstream of the intron's branch site in a sequence independent manner and may anchor the U2 snRNP to the pre-mRNA. Splicing factor 3b is also a component of the minor U12-type spliceosome. Subunit 3 has also been identified as a component of the STAGA (SPT3-TAF(II)31-GCN5L acetylase) transcription coactivator-HAT (histone acetyltransferase) complex and the TFTC (TATA-binding-protein-free TAF(II)-containing complex). These complexes may function in chromatin modification transcription splicing and DNA repair. Gene Aliases KIAA0017, RSE1, SAP130, SF3b130, STAF130 RefSeq Accession No. NC_000016.9, NG_027529.1, NT_010498.15 UniGene ID Hs.514435 Ensembl Gene ID ENSG00000189091 Entrez Gene ID 23450 Assay Information Unique Assay ID qHsaCIP0030454 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000302516, ENST00000310750 Amplicon Context Sequence GAGCCACTGGCATCAGCTTTGCCATTCATGGAAACTTTTCTGGAACCAAACAACA AGAAATTGTTGTTTCCCGTGGGAAGATCTTGGAGCTGCTTCGCCCAGACCCCAA CACTGGCAAAGTACATACCCTACTCACTGTGGAAGTATTCGGTGTTATCCGGTCA CTCATGGCCTTT Amplicon Length (bp) 146 Chromosome Location 16:70560588-70562909 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9986 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 18.72 cDNA Tm (Celsius) 83 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

The Biological Function and Clinical Significance of SF3B1 Mutations In
Zhou et al. Biomarker Research (2020) 8:38 https://doi.org/10.1186/s40364-020-00220-5 REVIEW Open Access The biological function and clinical significance of SF3B1 mutations in cancer Zhixia Zhou1* , Qi Gong2, Yin Wang1, Mengkun Li1, Lu Wang1, Hongfei Ding1 and Peifeng Li1* Abstract Spliceosome mutations have become the most interesting mutations detected in human cancer in recent years. The spliceosome, a large, dynamic multimegadalton small nuclear ribonucleoprotein composed of small nuclear RNAs associated with proteins, is responsible for removing introns from precursor mRNA (premRNA) and generating mature, spliced mRNAs. SF3B1 is the largest subunit of the spliceosome factor 3b (SF3B) complex, which is a core component of spliceosomes. Recurrent somatic mutations in SF3B1 have been detected in human cancers, including hematological malignancies and solid tumors, and indicated to be related to patient prognosis. This review summarizes the research progress of SF3B1 mutations in cancer, including SF3B1 mutations in the HEAT domain, the multiple roles and aberrant splicing events of SF3B1 mutations in the pathogenesis of tumors, and changes in mutated cancer cells regarding sensitivity to SF3B small-molecule inhibitors. In addition, the potential of SF3B1 or its mutations to serve as biomarkers or therapeutic targets in cancer is discussed. The accumulated knowledge about SF3B1 mutations in cancer provides critical insight into the integral role the SF3B1 protein plays in mRNA splicing and suggests new targets for anticancer therapy. Keywords: SF3B1, Mutation, Cancer, RNA splicing Background human diseases, including cancer [5, 6]. Indeed, genome- Of the 3.3 billion base pairs of haploid DNA in the human wide studies have revealed more than 15,000 tumor- genome, approximately 20,000 protein-coding genes have associated splice variants in a wide variety of cancers [7, 8]. -

Dramatically Reduced Spliceosome in Cyanidioschyzon Merolae
Dramatically reduced spliceosome in PNAS PLUS Cyanidioschyzon merolae Martha R. Starka, Elizabeth A. Dunnb, William S. C. Dunna, Cameron J. Grisdalec, Anthony R. Danielea, Matthew R. G. Halsteada, Naomi M. Fastc, and Stephen D. Radera,b,1 aDepartment of Chemistry, University of Northern British Columbia, Prince George, BC, V2N 4Z9 Canada; and Departments of bBiochemistry and Molecular Biology, and cBotany, University of British Columbia, Vancouver, BC, V6T 1Z4 Canada Edited by Joan A. Steitz, Howard Hughes Medical Institute, Yale University, New Haven, CT, and approved February 9, 2015 (received for review September 1, 2014) The human spliceosome is a large ribonucleoprotein complex that C. merolae is an acidophilic, unicellular red alga that grows at catalyzes pre-mRNA splicing. It consists of five snRNAs and more temperatures of up to 56 °C (6). At 16.5 million base pairs, its than 200 proteins. Because of this complexity, much work has genome is similar in size to that of S. cerevisiae and contains focusedontheSaccharomyces cerevisiae spliceosome, viewed as a comparable number of genes; however only one tenth as many a highly simplified system with fewer than half as many splicing introns were annotated in C. merolae: 26 intron-containing factors as humans. Nevertheless, it has been difficult to ascribe genes, 0.5% of the genome (6). The small number of introns in a mechanistic function to individual splicing factors or even to dis- C. merolae raises the questions of whether the full complexity cern which are critical for catalyzing the splicing reaction. We have of the canonical splicing machinery has been maintained or C merolae identified and characterized the splicing machinery from the red alga whether . -

Expression Levels of SF3B3 Correlate with Prognosis and This Is the Author's Manuscript of Article Published in Final Edited Form As
endocrine resistance in estrogen receptor-positive breast cancer. Modern Pathology, 28(5), 677–685. http://doi.org/10.1038/modpathol.2014.146 Gökmen-Polar, Y., Neelamraju, Goswami, C. P., Gu, X., Nallamothu, G., Janga, S. C., & Badve, (2015). Expression levels of SF3B3 correlate with prognosis and This is the author's manuscript of article published in final edited form as: 1 Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen 2 receptor positive breast cancer 3 4 Yesim Gökmen-Polar1*, Yaseswini Neelamraju3, Chirayu P. Goswami4, Xiaoping Gu1, 5 Gouthami Nallamothu1, Sarath Chandra Janga3,4,5, and Sunil Badve1,2,6* 6 Departments of 1Pathology and Laboratory Medicine, 2Medicine, Indiana University School of 7 Medicine, Indianapolis, IN, 3Department of Biohealth Informatics, School of Informatics and 8 Computing, IUPUI, Indianapolis, IN, 4Center for Computational Biology and Bioinformatics, 9 Indiana University School of Medicine; 5Department of Medical and Molecular Genetics, 10 Indiana University School of Medicine; 6Indiana University Melvin and Bren Simon Cancer 11 Center, Indianapolis, IN. 12 13 Grant Support 14 This work was supported by the Susan G Komen for the Cure® and 100 Voices of Hope to Sunil 15 Badve. 16 17 Disclosure of Potential Conflicts of Interest 18 The authors disclose no potential conflicts of interest. 19 20 *Address of corresponding: 21 Yesim Gökmen-Polar, PhD 22 Department of Pathology and Laboratory Medicine 23 Indiana University School of Medicine 24 635 Barnhill Dr., MS 0038 -

What Do the Structures of GCN5-Containing Complexes Teach Us About Their Function? Dominique Helmlinger, Gábor Papai, Didier Devys, Làszlo Tora
What do the structures of GCN5-containing complexes teach us about their function? Dominique Helmlinger, Gábor Papai, Didier Devys, Làszlo Tora To cite this version: Dominique Helmlinger, Gábor Papai, Didier Devys, Làszlo Tora. What do the structures of GCN5- containing complexes teach us about their function?. Biochimica et Biophysica Acta - Gene Regulatory Mechanisms , Elsevier, 2020, pp.194614. 10.1016/j.bbagrm.2020.194614. hal-02990243 HAL Id: hal-02990243 https://hal.archives-ouvertes.fr/hal-02990243 Submitted on 5 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License BBA - Gene Regulatory Mechanisms xxx (xxxx) xxxx Contents lists available at ScienceDirect BBA - Gene Regulatory Mechanisms journal homepage: www.elsevier.com/locate/bbagrm Review What do the structures of GCN5-containing complexes teach us about their ☆ function? ⁎ ⁎ Dominique Helmlingera, Gábor Papaib,c,d,e, Didier Devysb,c,d,e, , László Torab,c,d,e, a CRBM, University of Montpellier, CNRS, Montpellier, France b Institut de Génétique et de Biologie Moléculaire et Cellulaire, 67404 Illkirch, France c Centre National de la Recherche Scientifique (CNRS), UMR7104, 67404 Illkirch, France d Institut National de la Santé et de la Recherche Médicale (INSERM), U1258, 67404 Illkirch, France e Université de Strasbourg, 67404 Illkirch, France ARTICLE INFO ABSTRACT Keywords: Transcription initiation is a major regulatory step in eukaryotic gene expression. -

Alternative Splicing of EZH2 Pre-Mrna by SF3B3 Contributes To
Published OnlineFirst November 22, 2016; DOI: 10.1158/1078-0432.CCR-16-2020 Biology of Human Tumors Clinical Cancer Research Alternative Splicing of EZH2 pre-mRNA by SF3B3 Contributes to the Tumorigenic Potential of Renal Cancer Ke Chen1,2, Haibing Xiao1,2,3, Jin Zeng1,2, Gan Yu1,2, Hui Zhou1,2, Chunhua Huang4, Weimin Yao1,2, Wei Xiao1,2, Junhui Hu1,2, Wei Guan1,2, Lily Wu5, Jiaoti Huang6, Qihong Huang7,HuaXu1,2, and Zhangqun Ye1,2 Abstract Purpose: Deregulation or mutation of the EZH2 gene causes ited, and EZH2 promoted, cell growth, migration, proliferation, various tumors, including clear cell renal cell carcinoma (ccRCC). and tumorigenicity in a xenograft model. Mechanistic studies Although several splice variants of EZH2 have been identified, demonstrated that EZH2D14 isoform functions as a dominant- little is known about how EZH2 splicing is regulated or the negative inhibitor of full-length EZH2. Coexpression of EZH2D14 contribution of alternative splicing to its protumorigenic variant with full-length EZH2 not only abrogated DAB2IP and functions. HOXA9 suppression but also inhibited EZH2-driven tumorigen- Experimental Design: We conducted RT-PCR, Western blot esis. Strikingly, the splicing factor SF3B3 stimulates inclusion of analysis, and IHC techniques to examine EZH2 and its alternative exon14 and has pro-proliferative activity. Importantly, the upre- splicing transcript expression in renal cancer tissue and renal gulation of SF3B3 expression observed in clinical ccRCC samples cancer cell lines. Proliferation, migration, clonogenicity, and parallels the increased inclusion of EZH2 exon14, and the SF3B3 tumorigenicity of renal cancer cells either exhibiting knockdown level is associated with higher tumor stage and poor overall of EZH2 or its splicing factor SF3B3 were assessed by CCK8, survival. -

Dramatically Reduced Spliceosome in Cyanidioschyzon Merolae
Dramatically reduced spliceosome in PNAS PLUS Cyanidioschyzon merolae Martha R. Starka, Elizabeth A. Dunnb, William S. C. Dunna, Cameron J. Grisdalec, Anthony R. Danielea, Matthew R. G. Halsteada, Naomi M. Fastc, and Stephen D. Radera,b,1 aDepartment of Chemistry, University of Northern British Columbia, Prince George, BC, V2N 4Z9 Canada; and Departments of bBiochemistry and Molecular Biology, and cBotany, University of British Columbia, Vancouver, BC, V6T 1Z4 Canada Edited by Joan A. Steitz, Howard Hughes Medical Institute, Yale University, New Haven, CT, and approved February 9, 2015 (received for review September 1, 2014) The human spliceosome is a large ribonucleoprotein complex that C. merolae is an acidophilic, unicellular red alga that grows at catalyzes pre-mRNA splicing. It consists of five snRNAs and more temperatures of up to 56 °C (6). At 16.5 million base pairs, its than 200 proteins. Because of this complexity, much work has genome is similar in size to that of S. cerevisiae and contains focusedontheSaccharomyces cerevisiae spliceosome, viewed as a comparable number of genes; however only one tenth as many a highly simplified system with fewer than half as many splicing introns were annotated in C. merolae: 26 intron-containing factors as humans. Nevertheless, it has been difficult to ascribe genes, 0.5% of the genome (6). The small number of introns in a mechanistic function to individual splicing factors or even to dis- C. merolae raises the questions of whether the full complexity cern which are critical for catalyzing the splicing reaction. We have of the canonical splicing machinery has been maintained or C merolae identified and characterized the splicing machinery from the red alga whether .