Network-Based Metaanalysis Identifies HNF4A and PTBP1 As Longitudinally Dynamic Biomarkers for Parkinson’S Disease
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

SF3B3) and Sin3a Associated Protein 130 (SAP130
cells Communication Ambiguity about Splicing Factor 3b Subunit 3 (SF3B3) and Sin3A Associated Protein 130 (SAP130) Paula I. Metselaar 1,* , Celine Hos 1, Olaf Welting 1, Jos A. Bosch 2,3, Aletta D. Kraneveld 4 , Wouter J. de Jonge 1 and Anje A. Te Velde 1 1 Tytgat Institute for Liver and Intestinal Research, AGEM, Amsterdam UMC, University of Amsterdam, 1105BK Amsterdam, The Netherlands; [email protected] (C.H.); [email protected] (O.W.); [email protected] (W.J.d.J.); [email protected] (A.A.T.V.) 2 Department of Psychology, University of Amsterdam, 1018WS Amsterdam, The Netherlands; [email protected] 3 Department of Medical Psychology, Amsterdam UMC, University of Amsterdam, 1001NK Amsterdam, The Netherlands 4 Division of Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University, 3584CG Utrecht, The Netherlands; [email protected] * Correspondence: [email protected] Abstract: In 2020, three articles were published on a protein that can activate the immune system by binding to macrophage-inducible C-type lectin receptor (Mincle). In the articles, the protein was referred to as ‘SAP130, a subunit of the histone deacetylase complex.’ However, the Mincle ligand the authors aimed to investigate is splicing factor 3b subunit 3 (SF3B3). This splicing factor is unrelated to SAP130 (Sin3A associated protein 130, a subunit of the histone deacetylase-dependent Sin3A corepressor complex). The conclusions in the three articles were formulated for SF3B3, Citation: Metselaar, P.I.; Hos, C.; while the researchers used qPCR primers and antibodies against SAP130. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Supplementary Files for Understanding the Functional Impact of Copy Number Alterations in Breast Cancers Using a Network Modeling Approach
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2016 Supplementary files for Understanding the functional impact of copy number alterations in breast cancers using a network modeling approach Sriganesh Srihari1#, Murugan Kalimutho2#, Samir Lal3, Jitin Singla4, Dhaval Patel4, Peter T. Simpson3,5, Kum Kum Khanna2 and Mark A. Ragan1* 1Institute for Molecular Bioscience, The University of Queensland, St. Lucia, QLD 4072, Australia 2 QIMR-Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia 3 The University of Queensland, UQ Centre for Clinical Research, Brisbane, QLD 4029, Australia 4 Indian Institute of Technology Roorkee, Roorkee, Uttarakhand 247667, India 5 The University of Queensland, School of Medicine, Brisbane, QLD 4006, Australia Supplementary website: http://bioinformatics.org.au/tools-data/ under NetStrat Section 4.1 of main text (Materials and Methods) Choosing cut-off A pair of genes (proteins) could be co-expressed due to a number of reasons – for example, by being co- regulated by the same transcription factor or due to co-functioning in a complex via direct interactions. To identify trans-associated genes we are only interested in gene pairs (i.e. interactions in the network) whose co- expression associates with the CNA of one or both genes. In Equation 2 (main manuscript), since we compute the weighted sum of CNAs, those gene pairs which do not exhibit any CNAs (zero or low CNAs) would be assigned zero or low weights and hence will not be accounted for. In addition, we would also like to discount gene pairs from the network that show low co-expression values, by using an cut-off on the co-expression. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features
cancers Review Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features Valeria Visconte 1, Megan O. Nakashima 2 and Heesun J. Rogers 2,* 1 Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] 2 Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-445-2719 Received: 15 October 2019; Accepted: 19 November 2019; Published: 22 November 2019 Abstract: Components of the pre-messenger RNA splicing machinery are frequently mutated in myeloid malignancies. Mutations in LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2 genes occur at various frequencies ranging between 40% and 85% in different subtypes of myelodysplastic syndrome (MDS) and 5% and 10% of acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs). In some instances, splicing factor (SF) mutations have provided diagnostic utility and information on clinical outcomes as exemplified by SF3B1 mutations associated with increased ring sideroblasts (RS) in MDS-RS or MDS/MPN-RS with thrombocytosis. SF3B1 mutations are associated with better survival outcomes, while SRSF2 mutations are associated with a shorter survival time and increased AML progression, and U2AF1 mutations with a lower remission rate and shorter survival time. Beside the presence of mutations, transcriptomics technologies have shown that one third of genes in AML patients are differentially expressed, leading to altered transcript stability, interruption of protein function, and improper translation compared to those of healthy individuals. The detection of SF mutations demonstrates the importance of splicing abnormalities in the hematopoiesis of MDS and AML patients given the fact that abnormal splicing regulates the function of several transcriptional factors (PU.1, RUNX1, etc.) crucial in hematopoietic function. -

Splicing Factors Sf3a2 and Prp31 Have Direct Roles in Mitotic Chromosome Segregation
RESEARCH ARTICLE Splicing factors Sf3A2 and Prp31 have direct roles in mitotic chromosome segregation Claudia Pellacani1†, Elisabetta Bucciarelli1†, Fioranna Renda2‡, Daniel Hayward3, Antonella Palena1, Jack Chen3, Silvia Bonaccorsi2, James G Wakefield3, Maurizio Gatti1,2*, Maria Patrizia Somma1* 1Istituto di Biologia e Patologia Molecolari del CNR, Sapienza Universita` di Roma, Roma, Italy; 2Dipartimento di Biologia e Biotecnologie “C. Darwin”, Sapienza Universita` di Roma, Roma, Italy; 3Biosciences/Living Systems Institute, College of Life and Environmental Sciences, University of Exeter, Exeter, United Kingdom Abstract Several studies have shown that RNAi-mediated depletion of splicing factors (SFs) results in mitotic abnormalities. However, it is currently unclear whether these abnormalities reflect defective splicing of specific pre-mRNAs or a direct role of the SFs in mitosis. Here, we show that two highly conserved SFs, Sf3A2 and Prp31, are required for chromosome segregation in both Drosophila and human cells. Injections of anti-Sf3A2 and anti-Prp31 antibodies into Drosophila *For correspondence: embryos disrupt mitotic division within 1 min, arguing strongly against a splicing-related mitotic [email protected] (MG); function of these factors. We demonstrate that both SFs bind spindle microtubules (MTs) and the [email protected] Ndc80 complex, which in Sf3A2- and Prp31-depleted cells is not tightly associated with the (MPS) kinetochores; in HeLa cells the Ndc80/HEC1-SF interaction is restricted to the M phase. -

Download From
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.02.279679; this version posted September 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A high-throughput CRISPR interference screen for dissecting functional regulators of GPCR/cAMP signaling Khairunnisa Mentari Semesta1, Ruilin Tian2,3,4, Martin Kampmann2,3, Mark von Zastrow5,6 and Nikoleta G. Tsvetanova1* 1 Department of Pharmacology and Cancer Biology, Duke University, Durham, NC 27710, USA 2 Insitute for Neurodegenerative Diseases, Department of Biochemistry and Biophysics, University of California, San Francisco, CA 94158, USA 3 Chen-Zuckerberg Biohub, San Francisco, CA 94158, USA 4 Biophysics Graduate Program, University of California, San Francisco, CA 94158, USA 5 Department of Psychiatry, University of California, San Francisco, CA 94158, USA 6 Department of Cellular & Molecular Pharmacology, University of California, San Francisco, CA 94158, USA * Correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.09.02.279679; this version posted September 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Abstract 2 G protein-coupled receptors (GPCRs) allow cells to respond to chemical and sensory 3 stimuli through generation of second messengers, such as cyclic AMP (cAMP), which in 4 turn mediate a myriad of processes, including cell survival, proliferation, and 5 differentiation. -
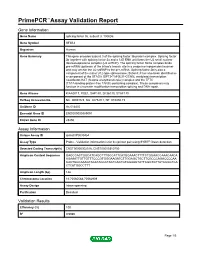
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name splicing factor 3b, subunit 3, 130kDa Gene Symbol SF3B3 Organism Human Gene Summary This gene encodes subunit 3 of the splicing factor 3b protein complex. Splicing factor 3b together with splicing factor 3a and a 12S RNA unit forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP). The splicing factor 3b/3a complex binds pre-mRNA upstream of the intron's branch site in a sequence independent manner and may anchor the U2 snRNP to the pre-mRNA. Splicing factor 3b is also a component of the minor U12-type spliceosome. Subunit 3 has also been identified as a component of the STAGA (SPT3-TAF(II)31-GCN5L acetylase) transcription coactivator-HAT (histone acetyltransferase) complex and the TFTC (TATA-binding-protein-free TAF(II)-containing complex). These complexes may function in chromatin modification transcription splicing and DNA repair. Gene Aliases KIAA0017, RSE1, SAP130, SF3b130, STAF130 RefSeq Accession No. NC_000016.9, NG_027529.1, NT_010498.15 UniGene ID Hs.514435 Ensembl Gene ID ENSG00000189091 Entrez Gene ID 23450 Assay Information Unique Assay ID qHsaCIP0030454 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000302516, ENST00000310750 Amplicon Context Sequence GAGCCACTGGCATCAGCTTTGCCATTCATGGAAACTTTTCTGGAACCAAACAACA AGAAATTGTTGTTTCCCGTGGGAAGATCTTGGAGCTGCTTCGCCCAGACCCCAA CACTGGCAAAGTACATACCCTACTCACTGTGGAAGTATTCGGTGTTATCCGGTCA CTCATGGCCTTT Amplicon Length (bp) 146 Chromosome Location 16:70560588-70562909 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9986 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 18.72 cDNA Tm (Celsius) 83 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase.