Diversity Patterns of Bacteriophages Infecting Aggregatibacter and Haemophilus Species Across Clades and Niches
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reporting of Diseases and Conditions Regulation, Amendment, M.R. 289/2014
THE PUBLIC HEALTH ACT LOI SUR LA SANTÉ PUBLIQUE (C.C.S.M. c. P210) (c. P210 de la C.P.L.M.) Reporting of Diseases and Conditions Règlement modifiant le Règlement sur la Regulation, amendment déclaration de maladies et d'affections Regulation 289/2014 Règlement 289/2014 Registered December 23, 2014 Date d'enregistrement : le 23 décembre 2014 Manitoba Regulation 37/2009 amended Modification du R.M. 37/2009 1 The Reporting of Diseases and 1 Le présent règlement modifie le Conditions Regulation , Manitoba Règlement sur la déclaration de maladies et Regulation 37/2009, is amended by this d'affections , R.M. 37/2009. regulation. 2 Schedules A and B are replaced with 2 Les annexes A et B sont remplacées Schedules A and B to this regulation. par les annexes A et B du présent règlement. Coming into force Entrée en vigueur 3 This regulation comes into force on 3 Le présent règlement entre en vigueur January 1, 2015, or on the day it is registered le 1 er janvier 2015 ou à la date de son under The Statutes and Regulations Act , enregistrement en vertu de Loi sur les textes whichever is later. législatifs et réglementaires , si cette date est postérieure. December 19, 2014 Minister of Health/La ministre de la Santé, 19 décembre 2014 Sharon Blady 1 SCHEDULE A (Section 1) 1 The following diseases are diseases requiring contact notification in accordance with the disease-specific protocol. Common name Scientific or technical name of disease or its infectious agent Chancroid Haemophilus ducreyi Chlamydia Chlamydia trachomatis (including Lymphogranuloma venereum (LGV) serovars) Gonorrhea Neisseria gonorrhoeae HIV Human immunodeficiency virus Syphilis Treponema pallidum subspecies pallidum Tuberculosis Mycobacterium tuberculosis Mycobacterium africanum Mycobacterium canetti Mycobacterium caprae Mycobacterium microti Mycobacterium pinnipedii Mycobacterium bovis (excluding M. -

Reportable Disease Surveillance in Virginia, 2013
Reportable Disease Surveillance in Virginia, 2013 Marissa J. Levine, MD, MPH State Health Commissioner Report Production Team: Division of Surveillance and Investigation, Division of Disease Prevention, Division of Environmental Epidemiology, and Division of Immunization Virginia Department of Health Post Office Box 2448 Richmond, Virginia 23218 www.vdh.virginia.gov ACKNOWLEDGEMENT In addition to the employees of the work units listed below, the Office of Epidemiology would like to acknowledge the contributions of all those engaged in disease surveillance and control activities across the state throughout the year. We appreciate the commitment to public health of all epidemiology staff in local and district health departments and the Regional and Central Offices, as well as the conscientious work of nurses, environmental health specialists, infection preventionists, physicians, laboratory staff, and administrators. These persons report or manage disease surveillance data on an ongoing basis and diligently strive to control morbidity in Virginia. This report would not be possible without the efforts of all those who collect and follow up on morbidity reports. Divisions in the Virginia Department of Health Office of Epidemiology Disease Prevention Telephone: 804-864-7964 Environmental Epidemiology Telephone: 804-864-8182 Immunization Telephone: 804-864-8055 Surveillance and Investigation Telephone: 804-864-8141 TABLE OF CONTENTS INTRODUCTION Introduction ......................................................................................................................................1 -

Identification of Pasteurella Species and Morphologically Similar Organisms
UK Standards for Microbiology Investigations Identification of Pasteurella species and Morphologically Similar Organisms Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 1 of 28 © Crown copyright 2015 Identification of Pasteurella species and Morphologically Similar Organisms Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 2 of 28 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Pasteurella species and Morphologically Similar Organisms Contents ACKNOWLEDGMENTS ......................................................................................................... -

Thesis Final
THESIS/DISSERTATION APPROVED BY 4-24-2020 Barbara J. O’Kane Date Barbara J. O’Kane, MS, Ph.D, Chair Margaret Jergenson Margret A. Jergenson, DDS Neil Norton Neil S. Norton, BA, Ph.D. Gail M. Jensen, Ph.D., Dean i COMPARISON OF PERIODONTIUM AMONG SUBJECTS TREATED WITH CLEAR ALIGNERS AND CONVENTIONAL ORTHODONTICS By: Mark S. Jones A THESIS Presented to the Faculty of The Graduate College at Creighton University In Partial Fulfillment of Requirements For the Degree of Master of Science in the Department of Oral Biology Under the Supervision of Dr. Marcelo Mattos Advising from: Dr. Margaret Jergenson, Dr. Neil S. Norton, and Dr. Barbara O’Kane Omaha, Nebraska 2020 i iii Abstract INTRODUCTION: With the wider therapeutic use of clear aligners the need to investigate the periodontal health status and microbiome of clear aligners’ patients in comparison with users of fixed orthodontic has arisen and is the objective of this thesis. METHODS: A clinical periodontal evaluation was performed, followed by professional oral hygiene treatment on a patient under clear aligner treatment, another under fixed orthodontics and two controls that never received any orthodontic therapy. One week after, supragingival plaque, swabs from the orthodontic devices, and saliva samples were collected from each volunteer for further 16s sequencing and microbiome analysis. RESULTS: All participants have overall good oral hygiene. However, our results showed increases in supragingival plaque, higher number of probing depths greater than 3mm, higher number of bleeding sites on probing, and a higher amount of gingival recession in the subject treated with fixed orthodontics. A lower bacterial count was observed colonizing the clear aligners, with less diversity than the other samples analyzed. -
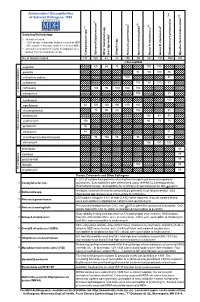
Antimicrobial Susceptibilities of Selected Pathogens, 1999
✔ Antimicrobial Susceptibilities * * † 7 of Selected Pathogens, 1999 8 e † a 2 ✔ ✔ † 3 ✔ † 4 5 6 culosis * r 1 urium spp. m L spp. Sampling Methodology 2 L † all isolates tested * ~ 20% sample of statewide isolates received at MDH spp. ~10% sample of statewide isolates received at MDH Salmonella ** all isolates tested from 7-county metropolitan area oup A streptococci ✔ oup B streptococci r isolates from a normally sterile site r Other (non-typhoidal) G Campylobacter Salmonella typhi Shigella Neisseria gonorrhoeae Neisseria meningitidis G Streptococcus pneumoni Mycobacterium tube No. of Isolates Tested 131 160 43 20 250 55 162 192 559 163 123456 123456% Susceptib123456le 123456123456 123456 123451623456 123451623456 ampicillin 1234561234566012345686 123456 15123456123456 100 100 123456123456 123451623451623451623451623456 123456 penicillin 123456123456123456123456123456 98100 100 76 123456 123456123456123456123456123456123456123456123456 123456 123451623451623451623456 123451623451623456 123456 cefuroxime sodium 123456123456123456123456 100123456123456123456 81 123456 123456123456123456123456123456123456123456123456 123456 cefotaxime 123451623451623451623451623456 100 100100 83 123456 123456123456123456123456123456 123456123456123456123456 123456 123456123456123456123456 ceftriaxone 123456 100 95 100 100 100 123451623451623451623456 -lactam antibiotics 123456123456123456123456123456 123456123456123456123456 β 123451623451623451623451623456 123451623456 123456 meropenem 123456123456123456123456123456 100 123456123456 83 123456 123456123456123456123456123456123456123456123456 -

Microbial NAD Metabolism: Lessons from Comparative Genomics
Dartmouth College Dartmouth Digital Commons Dartmouth Scholarship Faculty Work 9-2009 Microbial NAD Metabolism: Lessons from Comparative Genomics Francesca Gazzaniga Rebecca Stebbins Sheila Z. Chang Mark A. McPeek Dartmouth College Charles Brenner Carver College of Medicine Follow this and additional works at: https://digitalcommons.dartmouth.edu/facoa Part of the Biochemistry Commons, Genetics and Genomics Commons, Medicine and Health Sciences Commons, and the Microbiology Commons Dartmouth Digital Commons Citation Gazzaniga, Francesca; Stebbins, Rebecca; Chang, Sheila Z.; McPeek, Mark A.; and Brenner, Charles, "Microbial NAD Metabolism: Lessons from Comparative Genomics" (2009). Dartmouth Scholarship. 1191. https://digitalcommons.dartmouth.edu/facoa/1191 This Article is brought to you for free and open access by the Faculty Work at Dartmouth Digital Commons. It has been accepted for inclusion in Dartmouth Scholarship by an authorized administrator of Dartmouth Digital Commons. For more information, please contact [email protected]. MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS, Sept. 2009, p. 529–541 Vol. 73, No. 3 1092-2172/09/$08.00ϩ0 doi:10.1128/MMBR.00042-08 Copyright © 2009, American Society for Microbiology. All Rights Reserved. Microbial NAD Metabolism: Lessons from Comparative Genomics Francesca Gazzaniga,1,2 Rebecca Stebbins,1,2 Sheila Z. Chang,1,2 Mark A. McPeek,2 and Charles Brenner1,3* Departments of Genetics and Biochemistry and Norris Cotton Cancer Center, Dartmouth Medical School, Lebanon, New Hampshire -

The Old Testament Is Dying a Diagnosis and Recommended Treatment 1St Edition Download Free
THE OLD TESTAMENT IS DYING A DIAGNOSIS AND RECOMMENDED TREATMENT 1ST EDITION DOWNLOAD FREE Brent A Strawn | 9780801048883 | | | | | David T. Lamb Strawn offers a few other concrete suggestions about how to save the Old Testament, illustrating several of these by looking at the book of Deuteronomy as a model for second language acquisition. Retrieved 27 August The United States' Centers for Disease Control and Prevention CDC currently recommend that individuals who have been diagnosed and treated for gonorrhea avoid sexual contact with others until at least one week past the final day of treatment in order to prevent the spread of the bacterium. Brent Strawn reminds us of the Old Testament's important role in Christian faith and practice, criticizes current misunderstandings that contribute to its neglect, and offers ways to revitalize its use in the church. None, burning with urinationvaginal dischargedischarge from the penispelvic paintesticular pain [1]. Stunted language learners either: leave faith behind altogether; remain Christian, but look to other resources for how to live their lives; or balkanize in communities that prefer to speak something akin to baby talk — a pidgin-like form of the Old Testament and Bible as a whole — or, worse still, some sort of creole. Geoff, thanks for the reference. Log in. The guest easily identified the passage The Old Testament Is Dying A Diagnosis and Recommended Treatment 1st edition the New Testament, but the Old Testament passage was a swing, and a miss. Instead, our system considers things like how recent a review is and if the reviewer bought the item on Amazon. -

Product Sheet Info
Product Information Sheet for HM-206 Aggregatibacter aphrophilus, Oral Taxon immediately upon arrival. For long-term storage, the vapor phase of a liquid nitrogen freezer is recommended. Freeze- 545, Strain F0387 thaw cycles should be avoided. Catalog No. HM-206 Growth Conditions: Media: For research use only. Not for human use. Haemophilus Test medium or equivalent Chocolate agar or equivalent Contributor: Incubation: Jacques Izard, Assistant Member of the Staff, Department of Temperature: 37°C Molecular Genetics, The Forsyth Institute, Boston, Atmosphere: Aerobic with 5% CO2 Massachusetts, USA Propagation: 1. Keep vial frozen until ready for use, then thaw. Manufacturer: 2. Transfer the entire thawed aliquot into a single tube of broth. BEI Resources 3. Use several drops of the suspension to inoculate an agar slant and/or plate. Product Description: 4. Incubate the tube, slant and/or plate at 37°C for 24 to Bacteria Classification: Pasteurellaceae, Aggregatibacter 48 hours. Species: Aggregatibacter aphrophilus (formerly Haemophilus 1 aphrophilus) Citation: Subtaxon: Oral Taxon 545 Acknowledgment for publications should read “The following Strain: F0387 reagent was obtained through BEI Resources, NIAID, NIH as Original Source: Aggregatibacter aphrophilus (A. part of the Human Microbiome Project: Aggregatibacter aphrophilus), Oral Taxon 545, strain F0387 was isolated in aphrophilus, Oral Taxon 545, Strain F0387, HM-206.” 1984 from the subgingival dental plaque, at a healthy site, 2,3 of a 24-year-old female patient in the United States. Comments: A. aphrophilus, Oral Taxon 545, strain F0387 Biosafety Level: 1 (HMP ID 9335) is a reference genome for The Human Appropriate safety procedures should always be used with Microbiome Project (HMP). -

Bacterial Diversity and Functional Analysis of Severe Early Childhood
www.nature.com/scientificreports OPEN Bacterial diversity and functional analysis of severe early childhood caries and recurrence in India Balakrishnan Kalpana1,3, Puniethaa Prabhu3, Ashaq Hussain Bhat3, Arunsaikiran Senthilkumar3, Raj Pranap Arun1, Sharath Asokan4, Sachin S. Gunthe2 & Rama S. Verma1,5* Dental caries is the most prevalent oral disease afecting nearly 70% of children in India and elsewhere. Micro-ecological niche based acidifcation due to dysbiosis in oral microbiome are crucial for caries onset and progression. Here we report the tooth bacteriome diversity compared in Indian children with caries free (CF), severe early childhood caries (SC) and recurrent caries (RC). High quality V3–V4 amplicon sequencing revealed that SC exhibited high bacterial diversity with unique combination and interrelationship. Gracillibacteria_GN02 and TM7 were unique in CF and SC respectively, while Bacteroidetes, Fusobacteria were signifcantly high in RC. Interestingly, we found Streptococcus oralis subsp. tigurinus clade 071 in all groups with signifcant abundance in SC and RC. Positive correlation between low and high abundant bacteria as well as with TCS, PTS and ABC transporters were seen from co-occurrence network analysis. This could lead to persistence of SC niche resulting in RC. Comparative in vitro assessment of bioflm formation showed that the standard culture of S. oralis and its phylogenetically similar clinical isolates showed profound bioflm formation and augmented the growth and enhanced bioflm formation in S. mutans in both dual and multispecies cultures. Interaction among more than 700 species of microbiota under diferent micro-ecological niches of the human oral cavity1,2 acts as a primary defense against various pathogens. Tis has been observed to play a signifcant role in child’s oral and general health. -

Actinomyces Naeslundii and Aggregatibacter Aphrophilus Brain Abscess in an Adolescent
Arch Clin Med Case Rep 2019; 3 (6): 409-413 DOI: 10.26502/acmcr.96550112 Case Report Actinomyces Naeslundii and Aggregatibacter Aphrophilus Brain Abscess in an Adolescent Michael Croix1, Christopher Schwarz2, Ryan Breuer3,4, Amanda B. Hassinger3,4, Kunal Chadha5, Mark Daniel Hicar4,6 1Division of Internal Medicine and Pediatrics, University at Buffalo. Buffalo, New York, USA 2Division of Emergency Medicine, University at Buffalo. Buffalo, New York, USA 3Division of Pediatric Critical Care, John R. Oishei Children’s Hospital. Buffalo, New York, USA 4Department of Pediatrics, University at Buffalo. Buffalo, New York, USA 5Division of Pediatric Emergency Medicine, University at Buffalo. Buffalo, New York, USA 6Division of Pediatric Infectious Diseases, University at Buffalo. Buffalo, New York, USA *Corresponding Authors: Dr. Mark Daniel Hicar, Department of Pediatrics, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, 1001 Main Street, Buffalo, NY, 14203 USA, Tel: (716) 323-0150; Fax: (716) 888-3804; E-mail: [email protected] (or) [email protected] Dr. Michael Croix, Division of Internal Medicine and Pediatrics, 300 Linwood Ave, Buffalo, NY, 14209 USA, Tel: (217) 840-5750; Fax: (716) 888-3804; E-mail: [email protected] Received: 20 July 2019; Accepted: 02 August 2019; Published: 04 November 2019 Abstract We report the case of a child with a brain abscess from which Actinomyces naeslundii and Aggregatibacter aphrophilus were isolated. The is the first case describing A. naeslundii causing a brain abscess. This case highlights the association of these two organisms which may affect antibiotic choice and therapy length. Keywords: Brain; Abscess; Actinomyces; Aggregatibacter 1. Case Report A 13 year old male with no known past medical history initially presented to the Emergency Department with one week of headache, nausea, and vomiting. -

1 Supplementary Material a Major Clade of Prokaryotes with Ancient
Supplementary Material A major clade of prokaryotes with ancient adaptations to life on land Fabia U. Battistuzzi and S. Blair Hedges Data assembly and phylogenetic analyses Protein data set: Amino acid sequences of 25 protein-coding genes (“proteins”) were concatenated in an alignment of 18,586 amino acid sites and 283 species. These proteins included: 15 ribosomal proteins (RPL1, 2, 3, 5, 6, 11, 13, 16; RPS2, 3, 4, 5, 7, 9, 11), four genes (RNA polymerase alpha, beta, and gamma subunits, Transcription antitermination factor NusG) from the functional category of Transcription, three proteins (Elongation factor G, Elongation factor Tu, Translation initiation factor IF2) of the Translation, Ribosomal Structure and Biogenesis functional category, one protein (DNA polymerase III, beta subunit) of the DNA Replication, Recombination and repair category, one protein (Preprotein translocase SecY) of the Cell Motility and Secretion category, and one protein (O-sialoglycoprotein endopeptidase) of the Posttranslational Modification, Protein Turnover, Chaperones category, as annotated in the Cluster of Orthologous Groups (COG) (Tatusov et al. 2001). After removal of multiple strains of the same species, GBlocks 0.91b (Castresana 2000) was applied to each protein in the concatenation to delete poorly aligned sites (i.e., sites with gaps in more than 50% of the species and conserved in less than 50% of the species) with the following parameters: minimum number of sequences for a conserved position: 110, minimum number of sequences for a flank position: 110, maximum number of contiguous non-conserved positions: 32000, allowed gap positions: with half. The signal-to-noise ratio was determined by altering the “minimum length of a block” parameter. -

Haemophilus Influenzae Genome Evolution During Persistence in The
Haemophilus influenzae genome evolution during PNAS PLUS persistence in the human airways in chronic obstructive pulmonary disease Melinda M. Pettigrewa, Christian P. Ahearnb,c, Janneane F. Gentd, Yong Konge,f,g, Mary C. Gallob,c, James B. Munroh,i, Adonis D’Melloh,i, Sanjay Sethic,j,k, Hervé Tettelinh,i,1, and Timothy F. Murphyb,c,l,1,2 aDepartment of Epidemiology of Microbial Diseases, Yale School of Public Health, New Haven, CT 06510; bDepartment of Microbiology and Immunology, University at Buffalo, The State University of New York, Buffalo, NY 14203; cClinical and Translational Research Center, University at Buffalo, The State University of New York, Buffalo, NY 14203; dDepartment of Environmental Health Sciences, Yale School of Public Health, New Haven, CT 06510; eDepartment of Biostatistics, Yale School of Public Health, New Haven, CT 06510; fDepartment of Molecular Biophysics and Biochemistry, Yale School of Medicine, New Haven, CT 06510; gW.M. Keck Foundation Biotechnology Resource Laboratory, Yale School of Medicine, New Haven, CT 06510; hInstitute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD 21201; iDepartment of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD 21201; jDivision of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University at Buffalo, The State University of New York, Buffalo, NY 14203; kDepartment of Medicine, Veterans Affairs Western New York Healthcare System, Buffalo, NY 14215; and lDivision of Infectious Diseases, Department of Medicine, University at Buffalo, The State University of New York, Buffalo, NY 14203 Edited by Rino Rappuoli, GSK Vaccines, Siena, Italy, and approved February 27, 2018 (received for review November 10, 2017) Nontypeable Haemophilus influenzae (NTHi) exclusively colonize and adaptation cannot be accurately studied in vitro or in animal infect humans and are critical to the pathogenesis of chronic obstruc- models as a result of the unique physiological and immunological tive pulmonary disease (COPD).