Proteasome Activators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Protein Structure & Folding
6 Protein Structure & Folding To understand protein folding In the last chapter we learned that proteins are composed of amino acids Goal as a chemical equilibrium. linked together by peptide bonds. We also learned that the twenty amino acids display a wide range of chemical properties. In this chapter we will see Objectives that how a protein folds is determined by its amino acid sequence and that the three-dimensional shape of a folded protein determines its function by After this chapter, you should be able to: the way it positions these amino acids. Finally, we will see that proteins fold • describe the four levels of protein because doing so minimizes Gibbs free energy and that this minimization structure and the thermodynamic involves both making the most favorable bonds and maximizing disorder. forces that stabilize them. • explain how entropy (S) and enthalpy Proteins exhibit four levels of structure (H) contribute to Gibbs free energy. • use the equation ΔG = ΔH – TΔS The structure of proteins can be broken down into four levels of to determine the dependence of organization. The first is primary structure, the linear sequence of amino the favorability of a reaction on acids in the polypeptide chain. By convention, the primary sequence is temperature. written in order from the amino acid at the N-terminus (by convention • explain the hydrophobic effect and its usually on the left) to the amino acid at the C-terminus. The second level role in protein folding. of protein structure, secondary structure, is the local conformation adopted by stretches of contiguous amino acids. -

DNA Glycosylase Exercise - Levels 1 & 2: Answer Key
Name________________________ StarBiochem DNA Glycosylase Exercise - Levels 1 & 2: Answer Key Background In this exercise, you will explore the structure of a DNA repair protein found in most species, including bacteria. DNA repair proteins move along DNA strands, checking for mistakes or damage. DNA glycosylases, a specific type of DNA repair protein, recognize DNA bases that have been chemically altered and remove them, leaving a site in the DNA without a base. Other proteins then come along to fill in the missing DNA base. Learning objectives We will explore the relationship between a protein’s structure and its function in a human DNA glycosylase called human 8-oxoguanine glycosylase (hOGG1). Getting started We will begin this exercise by exploring the structure of hOGG1 using a molecular 3-D viewer called StarBiochem. In this particular structure, the repair protein is bound to a segment of DNA that has been damaged. We will first focus on the structure hOGG1 and then on how this protein interacts with DNA to repair a damaged DNA base. • To begin using StarBiochem, please navigate to: http://mit.edu/star/biochem/. • Click on the Start button Click on the Start button for StarBiochem. • Click Trust when a prompt appears asking if you trust the certificate. • In the top menu, click on Samples à Select from Samples. Within the Amino Acid/Proteins à Protein tab, select “DNA glycosylase hOGG1 w/ DNA – H. sapiens (1EBM)”. “1EBM” is the four character unique ID for this structure. Take a moment to look at the structure from various angles by rotating and zooming on the structure. -

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Proteasomes on the Chromosome Cell Research (2017) 27:602-603
602 Cell Research (2017) 27:602-603. © 2017 IBCB, SIBS, CAS All rights reserved 1001-0602/17 $ 32.00 RESEARCH HIGHLIGHT www.nature.com/cr Proteasomes on the chromosome Cell Research (2017) 27:602-603. doi:10.1038/cr.2017.28; published online 7 March 2017 Targeted proteolysis plays an this process, both in mediating homolog ligases, respectively, as RN components important role in the execution and association and in providing crossovers that impact crossover formation [5, 6]. regulation of many cellular events. that tether homologs and ensure their In the first of the two papers, Ahuja et Two recent papers in Science identify accurate segregation. Meiotic recom- al. [7] report that budding yeast pre9∆ novel roles for proteasome-mediated bination is initiated by DNA double- mutants, which lack a nonessential proteolysis in homologous chromo- strand breaks (DSBs) at many sites proteasome subunit, display defects in some pairing, recombination, and along chromosomes, and the multiple meiotic DSB repair, in chromosome segregation during meiosis. interhomolog interactions formed by pairing and synapsis, and in crossover Protein degradation by the 26S pro- DSB repair drive homolog association, formation. Similar defects are seen, teasome drives a variety of processes culminating in the end-to-end homolog but to a lesser extent, in cells treated central to the cell cycle, growth, and dif- synapsis by a protein structure called with the proteasome inhibitor MG132. ferentiation. Proteins are targeted to the the synaptonemal complex (SC) [3]. These defects all can be ascribed to proteasome by covalently ligated chains SC-associated focal protein complexes, a failure to remove nonhomologous of ubiquitin, a small (8.5 kDa) protein. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
UNIVERSITAT POLITÈCNICA DE CATALUNYA DEPARTAMENT D’ENGINYERIA QUÍMICA “CONFORMATIONAL PROPERTIES OF CONSTRAINED PROLINE ANALOGUES AND THEIR APPLICATION IN NANOBIOLOGY” Alejandra Flores Ortega Supervisors: Dr. Carlos Alemán Llansó and Dr. Jordi Casanovas Salas. th Barcelona, 27 January 2009 “Chance is a word void of sense; nothing can exist without a cause”. François-Marie Arouet, Voltaire “Imagination will often carry us to worlds that never were. But without it, we go nowhere”. Carl Sagan iii ACKNOWLEDGEMENTS I would like to acknowledge to Dr. Carlos Aleman and Dr. Jordi Cassanovas Salas for an interesting research theme, and scientific support. I gratefully acknowledge to Dr. David Zanuy for interesting suggestions and strong discussions, without their support this would be an unfulfilled task. Also I, would like to address my thanks to all my colleagues in my group and department, specially Elaine Armelin for assiting me in many different ways. I thank not only my friends, but also colleagues for helping me to overcome the stressful time, without whom it would have been difficult to cope up. I wish to express my gratefulness to my parents, specially to my mother, María Esther, for all his care, and support. Also I will like to thanks to my friends and specially Jesus, Merches, Laura y Arturo. My PhD thesis have been finished for all this support. I am greatly indepted to Dr. Ruth Nussinov at NCI, Dr. Carlos Cativiela at the University of Zaragoza and Ana I. Jiménez at the “Instituto de Ciencias de Materiales de Aragon” for a collaborative effort. I wish to thank all my colleague in the “Chimie et Biochimie Théoriques, Faculté des Sciences et Techniques” in Nancy France, I will be grateful to have worked with : Pr. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
research papers Detection of trans–cis flips and peptide-plane flips in protein structures ISSN 1399-0047 Wouter G. Touw,a* Robbie P. Joostenb and Gert Vrienda* aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Center, Geert Grooteplein-Zuid 26-28, 6525 GA Nijmegen, The Netherlands, and bDepartment of Biochemistry, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. *Correspondence e-mail: [email protected], Received 4 February 2015 [email protected] Accepted 27 April 2015 A coordinate-based method is presented to detect peptide bonds that need Edited by G. J. Kleywegt, EMBL–EBI, Hinxton, correction either by a peptide-plane flip or by a trans–cis inversion of the peptide England bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. Keywords: peptide conformation; cis peptide A few examples are highlighted for which a correction of the peptide-plane bond; structure validation; structure correction. geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK. 1. Introduction Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ! (CiÀ1—CiÀ1—Ni—Ci ) typically has values around 180 (trans)or0 (cis), although exceptions are possible (Berkholz et al., 2012). The Ci–1—Ci distance is around 3.81 A˚ in the trans conformation and around 2.94 A˚ in the cis conformation. -
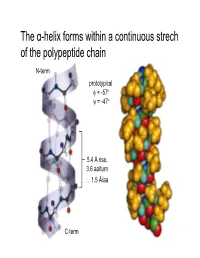
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Structure of the Lifeact–F-Actin Complex
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.16.951269; this version posted February 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Structure of the Lifeact–F-actin complex 2 Alexander Belyy1, Felipe Merino1,2, Oleg Sitsel1 and Stefan Raunser1* 3 1Department of Structural Biochemistry, Max Planck Institute of Molecular Physiology, Otto-Hahn-Str. 11, 44227 Dortmund, 4 Germany 5 2Current address: Department of Protein Evolution, Max Planck Institute for Developmental Biology, Max-Planck-Ring 5, 6 72076, Tübingen, Germany. 7 *Correspondence should be addressed to: [email protected] 8 9 Abstract 10 Lifeact is a short actin-binding peptide that is used to visualize filamentous actin (F-actin) 11 structures in live eukaryotic cells using fluorescence microscopy. However, this popular probe 12 has been shown to alter cellular morphology by affecting the structure of the cytoskeleton. The 13 molecular basis for such artefacts is poorly understood. Here, we determined the high- 14 resolution structure of the Lifeact–F-actin complex using electron cryo-microscopy. The 15 structure reveals that Lifeact interacts with a hydrophobic binding pocket on F-actin and 16 stretches over two adjacent actin subunits, stabilizing the DNase I-binding loop of actin in the 17 closed conformation. Interestingly, the hydrophobic binding site is also used by actin-binding 18 proteins, such as cofilin and myosin and actin-binding toxins, such as TccC3HVR from 19 Photorhabdus luminescens and ExoY from Pseudomonas aeruginosa.