Thesis Template
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Universidade Estadual De Campinas Instituto De Biologia
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA CAMPINAS 2016 1 VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA Dissertação apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do Título de Mestra em Biologia Funcional e Molecular na área de concentração de Bioquímica. Dissertation presented to the Institute of Biology of the University of Campinas in partial fulfillment of the requirements for the degree of Master in Functional and Molecular Biology, in the area of Biochemistry. ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA DISSERTAÇÃO DEFENDIDA PELA ALUNA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA E ORIENTADA PELO DANIEL MARTINS-DE-SOUZA. Orientador: Daniel Martins-de-Souza CAMPINAS 2016 2 Agência(s) de fomento e nº(s) de processo(s): CNPq, 151787/2F2014-0 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Saia-Cereda, Verônica Aparecida Monteiro, 1988- Sa21p O proteoma do corpo caloso da esquizofrenia / Verônica Aparecida Monteiro Saia Cereda. – Campinas, SP : [s.n.], 2016. Orientador: Daniel Martins de Souza. Dissertação (mestrado) – Universidade Estadual de Campinas, Instituto de Biologia. 1. Esquizofrenia. 2. Espectrometria de massas. 3. Corpo caloso. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Microenvironmental Reprogramming of Thymic Epithelial Cells to Skin Multipotent Stem Cells
Vol 466 | 19 August 2010 | doi:10.1038/nature09269 LETTERS Microenvironmental reprogramming of thymic epithelial cells to skin multipotent stem cells Paola Bonfanti1,2{, Ste´phanie Claudinot1,2, Alessandro W. Amici1,2, Alison Farley3, C. Clare Blackburn3 & Yann Barrandon1,2 The thymus develops from the third pharyngeal pouch of the or double positive cells (16%). After serial passaging, less than 2% of anterior gut and provides the necessary environment for thymo- the cells were positive for K8/18, while all were positive for K5/14, a poiesis (the process by which thymocytes differentiate into mature situation maintained in serial cultivation (Supplementary Fig. 1a). T lymphocytes) and the establishment and maintenance of self- These data indicate that clonogenic TECs originate from both the tolerance1–3. It contains thymic epithelial cells (TECs) that form a cortex and the medulla but that serial cultivation predominantly complex three-dimensional network organized in cortical and selected for a medullary phenotype. Clonogenic TECs were then medullary compartments, the organization of which is notably dif- ferent from simple or stratified epithelia4. TECs have an essential ab role in the generation of self-tolerant thymocytes through expres- sion of the autoimmune regulator Aire5,6, but the mechanisms involved in the specification and maintenance of TECs remain unclear7–9. Despite the different embryological origins of thymus and skin (endodermal and ectodermal, respectively), some cells of 10–12 the thymic medulla express stratified-epithelium markers , 4 105 79.3 15.1 10 interpreted as promiscuous gene expression. Here we show that 19.3 70.1 104 the thymus of the rat contains a population of clonogenic TECs 103 3 that can be extensively cultured while conserving the capacity to EGFP 10 EpCAM 102 integrate in a thymic epithelial network and to express major 102 0 histocompatibility complex class II (MHC II) molecules and Aire. -

Protein Expression Profiling in the Zebrafish (Danio Rerio) Embryos
Proteomics 2011, 11, 2003–2018 DOI 10.1002/pmic.201000442 2003 RESEARCH ARTICLE Protein expression profiling in the zebrafish (Danio rerio) embryos exposed to the microcystin-LR Guangyu Li1, Jun Chen1, Ping Xie1, Yan Jiang1, Laiyan Wu1 and Xuezhen Zhang 2 1 Donghu Experimental Station of Lake Ecosystems, State Key Laboratory for Freshwater Ecology and Biotechnology of China, Institute of Hydrobiology, The Chinese Academy of Sciences, Wuhan, P. R. China 2 Fisheries College of Huazhong Agricultural University, Wuhan, P. R. China Microcystin-leucine-arginine (MCLR) is the most toxic and the most commonly encountered Received: July 17, 2010 variant of microcystins (MCs) in aquatic environment, and it has the potential for develop- Revised: January 22, 2011 mental toxicity. A number of previous studies have described the developing toxicity of MCLR Accepted: February 1, 2011 based on conventional toxicological indices. However, the molecular mechanisms by which it expresses its toxicity during the early development remain largely unknown. To further our understanding of mechanisms of action and identify the potential protein biomarkers for MCLR exposure, a proteomic analysis was performed on developing zebrafish embryos exposed to 0.5 mg/L MCLR until 96 hours post-fertilization. 2-DE combined with MS was employed to detect and identify the protein profiles. Results showed that 75 spots from the 0.5 mg/L MCLR condition showed a significant increase or decrease in abundance compared with the control. In total, 40 proteins were identified. These proteins were mainly included in process related to oxidative stress, energetic metabolism, and the cytoskeleton assembly. MCLR exposure also affects the expression of the subunits of protein phosphatases 2A. -
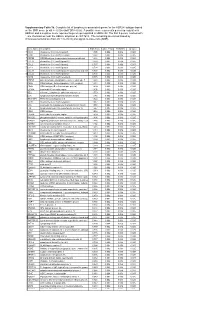
* Supplementary Table 3B. Complete List of Lymphocytic-Associated Genes
Supplementary Table 3b. Complete list of lymphocytic-associated genes for the HER2+I subtype based on the SNR score (p-val <= 0.005 and FDR<=0.05). A positive score represents genes up-regulated in HER2+I and a negative score represents genes up-regulated in HER2+NI. The first 9 genes, marked with *, are chemokines near the HER2+ amplicon at chr17q12. The remaining are sorted based by chromosomal location (from chr 1 to chr X) and signal-to-noise ratio (SNR). Gene Name Description SNR Score Feature P value FDR(BH) Q Value * CCL5 chemokine (C-C motif) ligand 5 1.395 0.002 0.036 0.025 * CCR7 chemokine (C-C motif) receptor 7 1.362 0.002 0.036 0.025 * CD79B CD79B antigen (immunoglobulin-associated beta) 1.248 0.002 0.036 0.025 * CCL13 chemokine (C-C motif) ligand 13 1.003 0.002 0.036 0.025 * CCL2 chemokine (C-C motif) ligand 2 0.737 0.004 0.056 0.037 * CCL8 chemokine (C-C motif) ligand 8 0.724 0.002 0.036 0.025 * CCL18 chemokine (C-C motif) ligand 18 (pulmonary and activ 0.703 0.002 0.036 0.025 * CCL23 chemokine (C-C motif) ligand 23 0.701 0.002 0.036 0.025 * CCR6 chemokine (C-C motif) receptor 6 0.715 0.002 0.036 0.025 PTPN7 protein tyrosine phosphatase, non-receptor type 7 1.861 0.002 0.036 0.025 CD3Z CD3Z antigen, zeta polypeptide (TiT3 complex) 1.811 0.002 0.036 0.025 CD48 CD48 antigen (B-cell membrane protein) 1.809 0.002 0.036 0.025 IL10RA interleukin 10 receptor, alpha 1.806 0.002 0.036 0.025 SELL selectin L (lymphocyte adhesion molecule 1) 1.773 0.002 0.036 0.025 LCK lymphocyte-specific protein tyrosine kinase 1.745 0.002 0.036 0.025 -

Supplementary Material
Supplementary material: Figure S1. Protein-Protein Interactions of the 127 Dysregulated proteins in brain tissues following IONP exposure. The graph was generated from STRING database. 125 nodes, 183 edges, 2.93 Average node degree, 0.411 Avg. local clustering coefficient, 122 Expected number of edges, PPI enrichment p-value 1.83e-07. 1 Figure S2. Protein-Protein Interactions of the 66 Dysregulated proteins in liver tissues following IONP exposure. The graph was generated from STRING database. 61 nodes, 103 edges, 3.38 Average node degree, 0.412 Avg. local clustering coefficient, 22 Expected number of edges, PPI enrichment p-value < 1.0e-16. 2 Figure S3. Protein-Protein Interactions of the 84 Dysregulated proteins in lung tissues following IONP exposure. The graph was generated from STRING database. 82 nodes, 125 edges, 3.05 Average node degree, 0.422 Avg. local clustering coefficient, 64 Expected number of edges, PPI enrichment p-value 1.07e-11. 3 Table S1. List of all dysregulated proteins in brain tissues following IONP exposure. A) Down regulated proteins and B) Upregulated proteins. Statistically significant iTRAQ ratios (p-value ratio and p-value sample≤0.05) for the 127 proteins that are dysregulated. A) Sequence Peptide Spectral P-Value P-Value Log 10 Accession Name Description Gene Coverage Ratio Count Count Ratio Sample Ratio % P00762 TRY1_RAT Anionic trypsin-1 Prss1 1 8 8.13 0.526 0.001 0.004 -0.279 P08592 A4_RAT Amyloid beta A4 protein App 1 2 1.17 0.546 0.010 0.004 -0.263 Q62696 DLG1_RAT Disks large homolog 1 Dlg1 1 1 1.32 -

Sox2 Promotes Malignancy in Glioblastoma by Regulating
Volume 16 Number 3 March 2014 pp. 193–206.e25 193 www.neoplasia.com Artem D. Berezovsky*, Laila M. Poisson†, Sox2 Promotes Malignancy in ‡ ‡ David Cherba , Craig P. Webb , Glioblastoma by Regulating Andrea D. Transou*, Nancy W. Lemke*, Xin Hong*, Laura A. Hasselbach*, Susan M. Irtenkauf*, Plasticity and Astrocytic Tom Mikkelsen*,§ and Ana C. deCarvalho* Differentiation1,2 *Department of Neurosurgery, Henry Ford Hospital, Detroit, MI; †Department of Public Health Sciences, Henry Ford Hospital, Detroit, MI; ‡Program of Translational Medicine, Van Andel Research Institute, Grand Rapids, MI; §Department of Neurology, Henry Ford Hospital, Detroit, MI Abstract The high-mobility group–box transcription factor sex-determining region Y–box 2 (Sox2) is essential for the maintenance of stem cells from early development to adult tissues. Sox2 can reprogram differentiated cells into pluripotent cells in concert with other factors and is overexpressed in various cancers. In glioblastoma (GBM), Sox2 is a marker of cancer stemlike cells (CSCs) in neurosphere cultures and is associated with the proneural molecular subtype. Here, we report that Sox2 expression pattern in GBM tumors and patient-derived mouse xenografts is not restricted to a small percentage of cells and is coexpressed with various lineage markers, suggesting that its expression extends beyond CSCs to encompass more differentiated neoplastic cells across molecular subtypes. Employing a CSC derived from a patient with GBM and isogenic differentiated cell model, we show that Sox2 knockdown in the differentiated state abolished dedifferentiation and acquisition of CSC phenotype. Furthermore, Sox2 deficiency specifically impaired the astrocytic component of a biphasic gliosarcoma xenograft model while allowing the formation of tumors with sarcomatous phenotype. -

BF0558-TUBA8 Antibody
Affinity Biosciences website:www.affbiotech.com order:[email protected] TUBA8 Antibody Cat.#: BF0558 Concn.: 1mg/ml Mol.Wt.: 50kDa Size: 50ul,100ul,200ul Source: Mouse Clonality: Monoclonal Application: ELISA 1:10000, WB 1:500-1:2000, IHC 1:200-1:1000, IF/ICC 1:200-1:1000, FCM 1:200-1:400 *The optimal dilutions should be determined by the end user. Reactivity: Human,Rat Purification: Affinity-chromatography. Specificity: TUBA8 antibody detects endogenous levels of total TUBA8. Immunogen: Purified recombinant fragment of human TUBA8 expressed in E. Coli. Uniprot: Q9NY65 Description: This gene encodes a member of the alpha tubulin protein family. Alpha tubulins are one of two core protein families (alpha and beta tubulins) that heterodimerize and assemble to form microtubules. Mutations in this gene are associated with polymicrogyria and optic nerve hypoplasia. Alternate splicing results in multiple transcript variants. Storage Condition and Mouse IgG1 in phosphate buffered saline (without Mg2+ and Buffer: Ca2+), pH 7.4, 150mM NaCl, 0.02% sodium azide and 50% glycerol.Store at -20 °C.Stable for 12 months from date of receipt. Figure 1: Western blot analysis using TUBA8 mAb against human TUBA8 (AA: 294-449) recombinant protein. (Expected MW is 50 kDa) Immunohistochemical analysis of paraffin-embedded medulla oblongata tissues using TUBA8 mouse mAb with DAB staining. 1 / 2 Affinity Biosciences website:www.affbiotech.com order:[email protected] Immunofluorescence analysis of Hela cells using TUBA8 mouse mAb (green). Blue: DRAQ5 fluorescent DNA dye. Red: Actin filaments have been labeled with Alexa Fluor-555 phalloidin. IMPORTANT: For western blot, incubate membrane with diluted primary Ab in 5% w/v milk , 1X TBS, 0.1% Tween®20 at 4°C with gentle shaking, overnight. -

ENCODE DCC Antibody Validation Document
ENCODE DCC Antibody Validation Document Date of Submission Name: Email: Lab Antibody Name: Target: Company/ Source: Catalog Number, database ID, laboratory Lot Number Antibody Description: Target Description: Species Target Species Host Validation Method #1 Validation Method #2 Purification Polyclonal/ Method Monoclonal Vendor URL: Reference (PI/ Publication Information) Please complete the following for antibodies to histone modifications: if your specifications are not listed in the drop-down box, please write-in the appropriate information Histone Name AA modified AA Position Modification Validation #1 Analysis Insert Validation Image (click here) Validation #2 Analysis Insert Validation Image (Click here) Results for lower band (~45 kDa): 1 >Keratin, type I cytoskeletal 10 n=1 Tax=Homo sapiens RepID=K1C10_HUMAN >keratin, type I cytoskeletal 10 n=1 Tax=Homo sapiens RepID=UPI00017BCE7F 2 >Keratin, type II cytoskeletal 2 epidermal n=1 Tax=Homo sapiens RepID=K22E_HUMAN 3 >Keratin, type II cytoskeletal 1 n=1 Tax=Homo sapiens RepID=K2C1_HUMAN 4 >Adipose most abundant gene transcript 2 protein n=2 Tax=Homo sapiens RepID=APM2_HUMAN 5 >Keratin, type I cytoskeletal 9 n=1 Tax=Homo sapiens RepID=K1C9_HUMAN 6 >Dermcidin isoform 2 n=1 Tax=Homo sapiens RepID=A5JHP3_HUMAN >DCD-1 n=3 Tax=Euarchontoglires RepID=DCD_HUMAN 7 >Trypsinogen n=1 Tax=Sus scrofa RepID=C5IWV5_PIG >Trypsin n=1 Tax=Sus scrofa RepID=TRYP_PIG 8 >Anti-human A33 heavy chain domain (Fragment) n=1 Tax=Oryctolagus cuniculus RepID=Q9N0W4_RABIT >Anti-human A33 heavy chain domain (Fragment) n=1 Tax=Oryctolagus cuniculus RepID=Q9N0W6_RABIT 9 >POU domain transcription factor OCT4-pg1 n=1 Tax=Homo sapiens RepID=D5K9V4_HUMAN >POU domain transcription factor OCT4-pg1 n=1 Tax=Homo sapiens RepID=D5K9W2_HUMAN 10 >Serum albumin n=1 Tax=Bos taurus RepID=ALBU_BOVIN >Serum albumin precursor (Allergen Bos d 6) (BSA). -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Identification of KRT16 As a Target of an Autoantibody Response in Complex Regional Pain Syndrome
Experimental Neurology 287 (2017) 14–20 Contents lists available at ScienceDirect Experimental Neurology journal homepage: www.elsevier.com/locate/yexnr Research Paper Identification of KRT16 as a target of an autoantibody response in complex regional pain syndrome Maral Tajerian, PhD a,b,c,⁎, Victor Hung, BSc a,c, Hamda Khan, BSc a,c, Lauren J Lahey, BSc a,c,d,YuanSun,PhDa,b,c, Frank Birklein, MD e, Heidrun H. Krämer, PhD f, William H Robinson, PhD a,c,d, Wade S Kingery, MD a,c,g, J David Clark, MD, PhD a,b,c a Veterans Affairs Palo Alto Health Care System Palo Alto, CA, USA b Department of Anesthesiology, Stanford University School of Medicine, Stanford, CA, USA c Palo Alto Veterans Institute for Research, Palo Alto, CA, USA d Division of Immunology and Rheumatology, Stanford University, Stanford, CA, USA e Department of Neurology, University Medical Center, Mainz, Germany f Department of Neurology, Justus Liebig University, Giessen, Germany g Physical Medicine and Rehabilitation Service, Veterans Affairs Palo Alto Health Care System, Palo Alto, CA, USA article info abstract Article history: Objective: Using a mouse model of complex regional pain syndrome (CRPS), our goal was to identify autoantigens Received 6 September 2016 in the skin of the affected limb. Received in revised form 12 October 2016 Methods: A CRPS-like state was induced using the tibia fracture/cast immobilization model. Three weeks after Accepted 18 October 2016 fracture, hindpaw skin was homogenized, run on 2-d gels, and probed by sera from fracture and control mice. Available online 20 October 2016 Spots of interest were analyzed by liquid chromatography-mass spectroscopy (LC-MS) and the list of targets val- idated by examining their abundance and subcellular localization.