Corine P. Kruiswijk Evolution of Major Histocompatibility
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biogeography and Evolution of the Carassius Auratus-Complex in East
Takada et al. BMC Evolutionary Biology 2010, 10:7 http://www.biomedcentral.com/1471-2148/10/7 RESEARCH ARTICLE Open Access Biogeography and evolution of the Carassius auratus-complex in East Asia Mikumi Takada1,2*, Katsunori Tachihara1, Takeshi Kon2, Gunji Yamamoto2, Kei’ichiro Iguchi3, Masaki Miya4, Mutsumi Nishida2 Abstract Background: Carassius auratus is a primary freshwater fish with bisexual diploid and unisexual gynogenetic triploid lineages. It is distributed widely in Eurasia and is especially common in East Asia. Although several genetic studies have been conducted on C. auratus, they have not provided clear phylogenetic and evolutionary descriptions of this fish, probably due to selection bias in sampling sites and the DNA regions analysed. As the first step in clarifying the evolutionary entity of the world’s Carassius fishes, we attempted to clarify the phylogeny of C. auratus populations distributed in East Asia. Results: We conducted a detailed analysis of a large dataset of mitochondrial gene sequences [CR, 323 bp, 672 sequences (528 sequenced + 144 downloaded); CR + ND4 + ND5 + cyt b, 4669 bp in total, 53 sequences] obtained from C. auratus in East Asia. Our phylogeographic analysis revealed two superlineages, one distributed mainly among the Japanese main islands and the other in various regions in and around the Eurasian continent, including the Ryukyus and Taiwan. The two superlineages include seven lineages with high regional specificity that are composed of endemic populations indigenous to each region. The divergence time of the seven lineages was estimated to be 0.2 million years ago (Mya) by a fossil-based method and 1.0-1.9 Mya by the molecular clock method. -
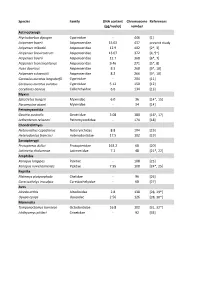
Table S2.Xlsx
Species Family DNA content Chromosome References (pg/nuclei) number Actinopterygii Ptychobarbus dipogon Cyprinidae ‐ 446 [1] Acipenser baerii Acipenseridae 15.02 437 present study Acipenser mikadoi Acipenseridae 12.9 402 [2*, 3] Acipenser brevirostrum Acipenseridae 13.07 372 [4, 5*] Acipenser baerii Acipenseridae 12.7 368 [6*, 7] Acipenser transmontanus Acipenseridae 9.46 271 [5*, 8] Huso dauricus Acipenseridae 8.3 268 [9*, 10] Acipenser schrenckii Acipenseridae 8.2 266 [9*, 10] Carassius auratus langsdorfii Cyprinidae ‐ 204 [11] Carassius auratus auratus Cyprinidae 5.12 150 [12] Corydoras aeneus Callichthyidae 6.6 134 [13] Myxini Eptatretus burgeri Myxinidae 6.0 36 [14*, 15] Paramyxine atami Myxinidae ‐ 34 [14] Petromyzontida Geotria australis Geotriidae 3.08 180 [16*, 17] Lethenteron reissneri Petromyzontidae ‐ 174 [18] Chondrichthyes Notorynchus cepedianus Notorynchidae 8.8 104 [19] Heterodontus francisci Heterodontidae 17.5 102 [19] Sarcopterygii Protopterus dolloi Protopteridae 163.2 68 [20] Latimeria chalumnae Latimeriidae 7.2 48 [21*, 22] Amphibia Xenopus longipes Pipidae ‐ 108 [23] Xenopus ruwenzoriensis Pipidae 7.95 108 [24*, 25] Reptilia Platemys platycephala Chelidae ‐ 96 [26] Carettochelys insculpta Carettochelyidae ‐ 68 [27] Aves Alcedo atthis Alcedinidae 2.8 138 [28, 29*] Upupa epops Upupidae 2.56 126 [28, 30*] Mammalia Tympanoctomys barrerae Octodontidae 16.8 102 [31, 32*] Ichthyomys pittieri Cricetidae ‐ 92 [33] References 1. Yu XY, Yu XJ. A schizothoracin fish species, Diptychus dipogon, with very high number of chromosomes Chrom Inform Serv. 1990;48:17-8. 2. Zhou H, Fujimoto T, Adachi S, Yamaha E, Arai K. Genome size variation estimated by flow cytometry in Acipenser mikadoi, Huso dauricus in relation to other species of Acipenseriformes. -

Polyphyletic Origin of Ornamental Goldfish
Food and Nutrition Sciences, 2015, 6, 1005-1013 Published Online August 2015 in SciRes. http://www.scirp.org/journal/fns http://dx.doi.org/10.4236/fns.2015.611104 Polyphyletic Origin of Ornamental Goldfish Aleksandr V. Podlesnykh1, Vladimir A. Brykov1,2, Lubov A. Skurikhina1 1Far Eastern Branch of the Russian Academy of Sciences, A. V. Zhirmunsky Institute of Marine Biology, Vladivostok, Russia 2School of Natural Sciences, Far Eastern Federal University, Vladivostok, Russia Email: [email protected] Received 6 May 2015; accepted 17 August 2015; published 20 August 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Mitochondrial DNA fragment of cytb was compared in all species Carassius auratus complex and three forms of ornamental goldfish. It is shown that the phylogenetic relationships between com- plex species correspond to the existing views, based on mtDNA data and geographical distribution. All forms of ornamental goldfish have a monophyletic origin from Chinese goldfish C. auratus au- ratus. The analysis showed that three nuclear genes (rps7, GH1 and Rh) in the two forms of orna- mental goldfish (Oriental twintail goldfish and Chinese Ranchu) were almost identical C. auratus auratus genes, while all three gene in another more simple form of goldfish (common goldfish) were highly homologous to carp Cyprinus carpio nuclear genes. The obtained data suggested that in the history of aquarium goldfish breeding occurred the stage of distant hybridization between goldfish and common carp. Subsequently, the nuclear genomes of some ornamental forms could be enriched by goldfish genes (a relatively recent form as Oriental twintail goldfish and Chinese Ranchu) or common carp genes (the simplest and most ancient forms like common goldfish) as a result of multidirectional breeding and selection of aquarium goldfish various forms. -

Electrophoretic Studies of Diploid, Triploid, and Tetraploid Forms of the Japanese Silver Crucian Carp, Carassius Auratus Langsdorfii
Japan.J.Ichthyol. 魚 類 学 雑 誌 40(1):65-75,1993 40(1):65-75,1993 Electrophoretic Studies of Diploid, Triploid, and Tetraploid Forms of the Japanese Silver Crucian Carp, Carassius auratus langsdorfii Yasushi Shimizu,1,2 Takashi Oshiro1 and Mitsuru Sakaizumi2,31 Department of Aquatic Biosciences, Tokyo University of Fisheries, 4-5-7 Konan, Minato-ku, Tokyo 108, Japan 2Department of Experimental Animal Science, The Tokyo Metropolitan Institute of Medical Science, 3-18-22 Honkomagome, Bunkyo-ku, Tokyo 113, Japan 3Present address: Department of Biology, College of General Education, Niigata University, 2-8050 Ikarashi, Niigata 950-21, Japan (Received October 29, 1992; in revisedform February 8, 1993; accepted March 22, 1993) Abstract The diploid-polyploid complex in the Japanese silver crucian carp (ginbuna), Carassius auratus langsdorfii, includes a diploid bisexual form, a triploid gynogenetic form and a tetraploid gynogenetic form. However, the origin of the polyploids remains to be clarified. Flow cytometric and electrophoretic analyses of Japanese crucian carp demonstrated that all the individuals with a two-banded pattern for an AMY-2 gene were polyploid, and 90% of the polyploids gave such a two-banded pattern. One of the two bands was not found in diploid subspecies in Japan but was present in both Korean crucian carp and goldfish. The diploid subspecies, kinbuna (C.auratus subsp.), which is distributed along the Pacific coast of eastern Japan, generated a diagnostic band of phosphoglucomutase. This band was also found in the polyploid "ginbuna" from the same region, but was observed in neither triploids nor diploids from other regions. -

Direct Cytotoxic Activity of CD8+ T Cells Against Ichthyophthirius Multifiliis
426 Abstracts / Fish & Shellfish Immunology 91 (2019) 421e472 importance for stress effects on the immune response in teleosts. Indi- # Corresponding author. vidual aspects of the interference of stress hormones (mainly cortisol) with E-mail address: [email protected] (J. Zou). immune processes have already been reported in some bony fish. Although less studied, the catecholamines adrenaline and noradrenaline have also shown to modulate the immune response of teleost leukocytes via a and b adrenergic receptors. This study aims to expand the actual knowledge on stress-induced immune modulation, in order to evaluate O-014. the effects of stress on the immune system of maraena whitefish (Cor- Direct cytotoxic activity of CD8þ T cells against Ichthyophthirius egonus maraena). This salmonid fish is highly sensitive to stress compared multifiliis in ginbuna crucian carp, Carassius auratus langsdorfii to other salmonid species long adapted to aquaculture. To this end, a large set of specific primers was designed for reverse-transcription quantitative Masaki Sukeda, Koumei Shiota, Takahiro Nagasawa, Miki Nakao, real-time PCR (RT-qPCR) analyses. The primer panel included cell-specific Tomonori Somamoto#. marker genes characterizing the distinct cell populations in the head kidney of C. maraena, which had been sorted using flow cytometry. In Laboratory of Marine Biochemistry, Department of Bioscience and addition, we analysed the expression of catecholamine and cortisol re- Biotechnology, Graduate School of Bioresource and Bioenvironmental ceptors in each population, in order to define the repertoire of stress- Sciences, Kyushu University, Fukuoka, Japan related modulators present in the cells. In the next step, we performed a series of in vitro stimulations of head kidney leukocytes to study the Abstract expression of genes involved in immune activation and acute phase A line of studies has shown that several humoral immune factors including together with catecholamine and cortisol receptors. -

In Vitro Characteristics of Cyprinid Herpesvirus 2: Effect of Kidney Extract Supplementation on Growth
Vol. 115: 223–232, 2015 DISEASES OF AQUATIC ORGANISMS Published August 20 doi: 10.3354/dao02885 Dis Aquat Org In vitro characteristics of cyprinid herpesvirus 2: effect of kidney extract supplementation on growth Tomoya Shibata1, Azusa Nanjo1, Masato Saito1, Keisuke Yoshii1, Takafumi Ito2, Teruyuki Nakanishi3, Takashi Sakamoto1, Motohiko Sano1,* 1Faculty of Marine Science, Tokyo University of Marine Science and Technology, Minato, Tokyo 108-8477, Japan 2Aquatic Animal Health Division, National Research Institute of Aquaculture, Fisheries Research Agency, Tamaki, Mie 519-0423, Japan 3Department of Veterinary Medicine, Nihon University, Fujisawa, Kanagawa 252-0880, Japan ABSTRACT: Herpesviral hematopoietic necrosis caused by goldfish hematopoietic necrosis virus (now identified as cyprinid herpesvirus 2, CyHV-2) has contributed to economic losses in goldfish Carassius auratus culture and is becoming a major obstacle in Prussian carp C. gibelio aquacul- ture in China. Several reports have described difficulties in culturing the virus, with the total loss of infectivity within several passages in cell culture. We succeeded in propagating CyHV-2 with a high infectious titer in a RyuF-2 cell line newly derived from the fin of the Ryukin goldfish variety using culture medium supplemented with 0.2% healthy goldfish kidney extract. The addition of kidney extract to the medium enabled rapid virus growth, resulting in the completion of cyto- pathic effect (CPE) within 4 to 6 d at 25°C. The extract also enabled reproducible virus culture 5−6 −1 with a titer of 10 TCID50 ml . The virus cultured using this protocol showed pathogenicity in goldfish after intraperitoneal injection. The virus grew in RyuF-2 cells at 15, 20, 25, 30, and 32°C but not at 34°C or higher. -

This Article Appeared in a Journal Published by Elsevier. the Attached
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/authorsrights Author's personal copy Molecular Immunology 70 (2016) 1–7 Contents lists available at ScienceDirect Molecular Immunology j ournal homepage: www.elsevier.com/locate/molimm Functional analysis of membrane-bound complement regulatory protein on T-cell immune response in ginbuna crucian carp a,b a,c a a Indriyani Nur , Nevien K. Abdelkhalek , Shiori Motobe , Ryota Nakamura , a a,∗ a Masakazu Tsujikura , Tomonori Somamoto , Miki Nakao a Laboratory of Marine Biochemistry, Department of Bioscience and Biotechnology, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, Fukuoka 812-8581, Japan b Aquaculture Department, Fisheries and Marine Science Faculty, Halu Oleo University, Kendari 93232, Indonesia c Department of Internal Medicine, Infectious and Fish diseases, Faculty of Veterinary Medicine, El-Mansoura University, Egypt a r t i c l e i n f o a b s t r a c t Article history: Complements have long been considered to be a pivotal component in innate immunity. Recent Received 9 August 2015 researches, however, highlight novel roles of complements in T-cell-mediated adaptive immunity. -

Spontaneous Triploidy in the California Roach Hesperoleucus Symmetricus (Pisces: Cyprinidae)
Cytogcnet. Cell Genet. 17: 144-149 (1976) Spontaneous triploidy in the California roach Hesperoleucus symmetricus (Pisces: Cyprinidae) J.R.G old 1 and J.C. A vise2 'Genetics Section, Texas A&M University, College Station, Tex., and "Department of Zoology, University of Georgia, Athens, Ga. Abstract A single triploid individual (3n = 75) of the California roach,Hesperoleucus symmetricus, was identified among a sample of nine specimens from the Russian River, California. The diploid number ofH. symmetricus, as revealed by the karyotypes of the remaining eight specimens, is 50. Aside from the all-female triploid unisexual fishes, this is the first report of a triploid fish from the wild, and the second report of a triploid in a bisexual fish species. The most likely origin of the triploid was probably fusion of a haploid sperm with an unreduced ovum. Among the vertebrates, viable triploids are exceedingly infrequent in nature, and, with very few exceptions, they have been found only in certain unisexual forms among the fishes(Schultz, 1971) and salamanders (M aslin , 1971). The exceptions include a few isolated individuals of the bird Gallus domesticus(O hno et al., 1963; Sarvei.la , 1970; A bdel - H ameed and Shoefner , 1971), a few species of salamanders and frogs (references in Cuellar and U yeno , 1972), and the fishSalmo gairdneri (C uellar and U yeno , 1972). Triploidy or diploid/triploid chimerism has been reported in other vertebrates, e.g., man, but is associated with embryonic mortality or early death(C hu et al., 1964; C arr , 1970; Schindler and M ikamo , 1970; Bloom , 1972). -

Morphological Identification of Haematopoietic Cells in Pronephros
Volume 60(2):113-118, 2016 Acta Biologica Szegediensis http://www2.sci.u-szeged.hu/ABS ARTICLE Morphological identification of haematopoietic cells in pronephros of common carp (Cyprinus carpio Linnaeus, 1758) Damir Suljeviæ, Erna Islamagić, Andi Alijagić*, Muhamed Fočak, Maja Mitrašinović-Brulić Department of Biology (Biochemistry and Physiology), Faculty of Science, University of Sarajevo, Sarajevo, Bosnia and Herzegovina ABSTRACT Haemopoietic tissue of the common carp pronephros (Cyprinus carpio Linnaeus, KEY WORDS 1758) was studied in regard of morphometric analysis of the erythropoietic, leukopoietic and common carp thrombopoietic cell populations. The aim of this study was to perform evaluation of immature haematopoiesis precursor cells in head kidney of common carp (sampled from Bardaca lake) because there is pronephros no well known data regarding this issue. Microscopic identification of haematopoietic cells cell-lineage included measurement of cell and nuclear size, nuclear-cytoplasmic ratio, determination of the cell area cell and nuclear shape, cytosol coloration and presence of specific granules in the cytosol. The frequency of immature cells and their cell area was also analyzed. Erythroblasts were the most abundant among all observed haemopoietic cell lineages and were the most variable in the size. The largest area was characteristic of monocyte precursors and no significant differences were observed regarding the cell area between prothrombocytes and lymphoblasts, which makes difficult in cell characterization. High number of emerged cells in short time also makes difficult to identify particular stages of maturation in some bloodlines. Rapid maturation of granuloid cells observed within the haemopoietic tissue indicates their functional significance in adaptation to the changeable microenvironment. Acta Biol Szeged 60(2):113-118 (2016) Introduction intestine (Stosik and Deptula 1993). -

Hexaploidy in Yellowfish Species (Barbus, Pisces, Cyprinidae) from Southern Africa
Journal of Fish Biology (1990) 37, 105-1 15 Hexaploidy in yellowfish species (Barbus, Pisces, Cyprinidae) from southern Africa L. K. OELLERMANNAND P. H. SICELTON* J.L.B. Smith Institute of Ichthyology, Private Bag 1015, Grahamstown 6140, Republic of South Africa (Received 18 July 1989, Accepted I February 1990) Five small-scaled yellowfish (large Burbus spp.) from southern Africa are shown to have modal 148 or 150chromosomes. Themajority ofcyprinid species have 2N = 50chromosomes, indicating that the yellowfish karyotype is hexaploid in origin. However, as there is no indication that the species are unisexual or that normal reproduction occurs by any means other than bisexual fertilization, the yellowfish karyotype is considered to have reverted to a diploid condition. Key words: karyology; yellowfish; Burbus; hexaploidy; southern Africa. I. INTRODUCTION The name ' yellowfish ' is given to several large-sized Burbus species in southern Africa (Jubb, 1967). The yellowfish fall into two groups on the basis of scale size. The group with smaller scales consists of five recognized species distributed in the Orange River system and adjacent drainage systems (Skelton, 1986). The second group, with larger scales, consists of two species, Barbus rnarequensis Smith, 1841 and Barbus codringtonii Boulenger, 1908, distributed in east coastal drainages from the Phongola River to the Zambezi River system. This study concerns the five small-scaled yellowfish species, viz. Barbus cupensis Smith, 1841 from the Olifants River system, Burbus aeneus (Burchell, 1822) and Burbus kimberleyensis Gilchrist & Thompson, 1913 from the Orange River system, Barbus polylepis Boulenger, 1907 from the Limpopo, lncomati and Phongola River systems, and Burbus natalensis Castelnau, 1861 from the rivers of Natal. -

Black, Grey and Watch Lists of Alien Species in the Czech Republic
A peer-reviewed open-access journal NeoBiota 28: 1–37 (2016)Black, Grey and Watch Lists of alien species in the Czech Republic... 1 doi: 10.3897/neobiota.28.4824 RESEARCH ARTICLE NeoBiota http://neobiota.pensoft.net Advancing research on alien species and biological invasions Black, Grey and Watch Lists of alien species in the Czech Republic based on environmental impacts and management strategy Jan Pergl1, Jiří Sádlo1, Adam Petrusek2, Zdeněk Laštůvka3, Jiří Musil4, Irena Perglová1, Radek Šanda5, Hana Šefrová6, Jan Šíma7, Vladimír Vohralík8, Petr Pyšek1,2 1 Institute of Botany, Department of Invasion Ecology, The Czech Academy of Sciences, CZ-252 43 Průhonice, Czech Republic 2 Department of Ecology, Faculty of Science, Charles University in Prague, Viničná 7, CZ-128 44 Praha 2, Czech Republic 3 Department of Zoology, Fisheries, Hydrobiology and Apidology, Mendel University in Brno, Zemědělská 1, CZ-613 00 Brno, Czech Republic 4 T.G. Masaryk Water Research Institute, Department of Aquatic Ecology, Podbabská 30, CZ-60 00 Prague 6, Czech Republic 5 National Museum, Department of Zoology, Václavské náměstí 68, CZ-115 79 Prague 1, Czech Republic 6 Department of Crop Science, Breeding and Plant Medicine, Mendel University in Brno, Zemědělská 1, CZ-613 00 Brno, Czech Republic 7 Ministry of the Environment of the Czech Republic, Department of Species Protection and Implementation of International Commitments, Vršovická 1442/65, CZ-100 10 Praha 10, Czech Republic 8 Department of Zoology, Faculty of Science, Charles University in Prague, Viničná 7, CZ-128 44 Praha 2, Czech Republic Corresponding author: Jan Pergl ([email protected]) Academic editor: I. -

Cytogenetic Characteristics of Cyprinidae Between Diploid and Spontaneous Triploid in Major River of Korea
한국해양생명과학회지[Journal of Marine Life Science] June 2018; 3(1): 9-21 JMLS http://jmls.or.kr Cytogenetic Characteristics of Cyprinidae between Diploid and Spontaneous Triploid in Major River of Korea In Bon Goo1, Sang Gu Lim2, Hyun Woo Gil3, In-Seok Park4*, Cheol Young Choi4 1Inland Aquaculture Research Center, National Institute of Fisheries Science (NIFS), Jinhae 51688, Korea 2Aquafeed Research Center, NIFS, Pohang 37517, Korea 3Bio-Monitoring Center, Sejong 30121, Korea 4 Division of Marine Bioscience, College of Ocean Science and Technology, Korea Maritime and Ocean University, Busan 49112, Korea Corresponding Author This study investigated cytogenetic and hematological and histological characteristics In-Seok Park between diploid and spontaneous triploid on Cyprinidae (Crucian carp, Carassius auratus; crucian carp, C. cuvieri and common carp, Cyprinus carpio) in four major rivers of Korea. Division of Marine Bioscience, College of In our results, DNA contents of triploid Cyprinidae were 50% more than those of diploid Ocean Science and Technology, Korea Cyprinidae. Also, erythrocyte size of triploid Cyprinidae was 50% larger than those of Maritime and Ocean University, Busan diploid Cyprinidae. In all sampling rivers, sex ratios of C. auratus were biased to female, 49112, Korea and especially, triploid groups of C. auratus were all female groups (p<0.05). In principle, E-mail : [email protected] sex ratios of C. cuvieri and common carp were equivalent between male and female. Keywords: Carassius auratus, C. cuvieri, Cyprinus carpio, Cytogenetic characteristics, Received : February 05, 2018 Spontaneous triploid Revised : May 09, 2018 Accepted : May 10, 2018 Introduction logical differences resulting from environmental influences (Nam et al., 1989).