Biogeography and Evolution of the Carassius Auratus-Complex in East
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
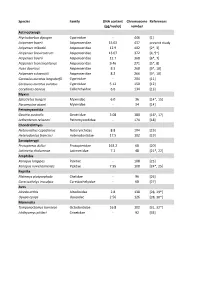
Table S2.Xlsx
Species Family DNA content Chromosome References (pg/nuclei) number Actinopterygii Ptychobarbus dipogon Cyprinidae ‐ 446 [1] Acipenser baerii Acipenseridae 15.02 437 present study Acipenser mikadoi Acipenseridae 12.9 402 [2*, 3] Acipenser brevirostrum Acipenseridae 13.07 372 [4, 5*] Acipenser baerii Acipenseridae 12.7 368 [6*, 7] Acipenser transmontanus Acipenseridae 9.46 271 [5*, 8] Huso dauricus Acipenseridae 8.3 268 [9*, 10] Acipenser schrenckii Acipenseridae 8.2 266 [9*, 10] Carassius auratus langsdorfii Cyprinidae ‐ 204 [11] Carassius auratus auratus Cyprinidae 5.12 150 [12] Corydoras aeneus Callichthyidae 6.6 134 [13] Myxini Eptatretus burgeri Myxinidae 6.0 36 [14*, 15] Paramyxine atami Myxinidae ‐ 34 [14] Petromyzontida Geotria australis Geotriidae 3.08 180 [16*, 17] Lethenteron reissneri Petromyzontidae ‐ 174 [18] Chondrichthyes Notorynchus cepedianus Notorynchidae 8.8 104 [19] Heterodontus francisci Heterodontidae 17.5 102 [19] Sarcopterygii Protopterus dolloi Protopteridae 163.2 68 [20] Latimeria chalumnae Latimeriidae 7.2 48 [21*, 22] Amphibia Xenopus longipes Pipidae ‐ 108 [23] Xenopus ruwenzoriensis Pipidae 7.95 108 [24*, 25] Reptilia Platemys platycephala Chelidae ‐ 96 [26] Carettochelys insculpta Carettochelyidae ‐ 68 [27] Aves Alcedo atthis Alcedinidae 2.8 138 [28, 29*] Upupa epops Upupidae 2.56 126 [28, 30*] Mammalia Tympanoctomys barrerae Octodontidae 16.8 102 [31, 32*] Ichthyomys pittieri Cricetidae ‐ 92 [33] References 1. Yu XY, Yu XJ. A schizothoracin fish species, Diptychus dipogon, with very high number of chromosomes Chrom Inform Serv. 1990;48:17-8. 2. Zhou H, Fujimoto T, Adachi S, Yamaha E, Arai K. Genome size variation estimated by flow cytometry in Acipenser mikadoi, Huso dauricus in relation to other species of Acipenseriformes. -

Japan Geoscience Union Meeting 2009 Presentation List
Japan Geoscience Union Meeting 2009 Presentation List A002: (Advances in Earth & Planetary Science) oral 201A 5/17, 9:45–10:20, *A002-001, Science of small bodies opened by Hayabusa Akira Fujiwara 5/17, 10:20–10:55, *A002-002, What has the lunar explorer ''Kaguya'' seen ? Junichi Haruyama 5/17, 10:55–11:30, *A002-003, Planetary Explorations of Japan: Past, current, and future Takehiko Satoh A003: (Geoscience Education and Outreach) oral 301A 5/17, 9:00–9:02, Introductory talk -outreach activity for primary school students 5/17, 9:02–9:14, A003-001, Learning of geological formation for pupils by Geological Museum: Part (3) Explanation of geological formation Shiro Tamanyu, Rie Morijiri, Yuki Sawada 5/17, 9:14-9:26, A003-002 YUREO: an analog experiment equipment for earthquake induced landslide Youhei Suzuki, Shintaro Hayashi, Shuichi Sasaki 5/17, 9:26-9:38, A003-003 Learning of 'geological formation' for elementary schoolchildren by the Geological Museum, AIST: Overview and Drawing worksheets Rie Morijiri, Yuki Sawada, Shiro Tamanyu 5/17, 9:38-9:50, A003-004 Collaborative educational activities with schools in the Geological Museum and Geological Survey of Japan Yuki Sawada, Rie Morijiri, Shiro Tamanyu, other 5/17, 9:50-10:02, A003-005 What did the Schoolchildren's Summer Course in Seismology and Volcanology left 400 participants something? Kazuyuki Nakagawa 5/17, 10:02-10:14, A003-006 The seacret of Kyoto : The 9th Schoolchildren's Summer Course inSeismology and Volcanology Akiko Sato, Akira Sangawa, Kazuyuki Nakagawa Working group for -

Zootaxa, Japanese Pseudosmittia Edwards (Diptera: Chironomidae)
Zootaxa 1198: 21–51 (2006) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA 1198 Copyright © 2006 Magnolia Press ISSN 1175-5334 (online edition) Japanese Pseudosmittia Edwards (Diptera: Chironomidae) OLE A. SÆTHER The Natural History Collections, Bergen Museum, University of Bergen, N-5020 Bergen, Norway. E-mail: [email protected] Abstract The types of species previously placed in Pseudosmittia Edwards and some related genera in the Sasa collection at The National Museum of Sciences, Tokyo, Japan, have been examined. Twenty- four new synonyms are given: Pseudosmittia ogasatridecima Sasa et Suzuki, 1997a is a synonym of P. bifurcata (Tokunaga, 1936); P. jintuvicesima Sasa, 1996, and P. seiryupequea Sasa, Suzuki et Sakai, 1998 of P. danconai (Marcuzzi, 1947); P. mongolzeaea Sasa et Suzuki, 1997b of P. f orc ipa ta (Goetghebuer, 1921); P. hachijotertia Sasa, 1994 of P. holsata Thienemann et Strenzke, 1940; P. itachibifurca Sasa et Kawai, 1987, P. furudobifurca Sasa et Arakawa, 1994, P. hibaribifurca Sasa, 1993, and P. (Nikismittia) shofukuundecima Sasa, 1998 of P. mathildae Albu, 1968; P. yakymenea Sasa et Suzuki, 2000a, and P. yakyneoa Sasa et Suzuki, 2000a of P. nishiharaensis Sasa et Hasegawa, 1988; P. kurobeokasia Sasa et Okazawa, 1992a, P. togarisea Sasa et Okazawa, 1992b, P. hachijosecunda Sasa, 1994, P. to ya m a re s e a Sasa, 1996, P. yakyopea Sasa et Suzuki, 2000a, P. yakypequea Sasa et Suzuki, 2000a, Parakiefferiella hidakagehea Sasa et Suzuki, 2000b, and Parakiefferiella hidakaheia Sasa et Suzuki, 2000b of Pseudosmittia oxoniana (Edwards, 1922); P. famikelea Sasa, 1996a of P. tokaraneoa Sasa et Suzuki, 1995; P. -

Polyphyletic Origin of Ornamental Goldfish
Food and Nutrition Sciences, 2015, 6, 1005-1013 Published Online August 2015 in SciRes. http://www.scirp.org/journal/fns http://dx.doi.org/10.4236/fns.2015.611104 Polyphyletic Origin of Ornamental Goldfish Aleksandr V. Podlesnykh1, Vladimir A. Brykov1,2, Lubov A. Skurikhina1 1Far Eastern Branch of the Russian Academy of Sciences, A. V. Zhirmunsky Institute of Marine Biology, Vladivostok, Russia 2School of Natural Sciences, Far Eastern Federal University, Vladivostok, Russia Email: [email protected] Received 6 May 2015; accepted 17 August 2015; published 20 August 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Mitochondrial DNA fragment of cytb was compared in all species Carassius auratus complex and three forms of ornamental goldfish. It is shown that the phylogenetic relationships between com- plex species correspond to the existing views, based on mtDNA data and geographical distribution. All forms of ornamental goldfish have a monophyletic origin from Chinese goldfish C. auratus au- ratus. The analysis showed that three nuclear genes (rps7, GH1 and Rh) in the two forms of orna- mental goldfish (Oriental twintail goldfish and Chinese Ranchu) were almost identical C. auratus auratus genes, while all three gene in another more simple form of goldfish (common goldfish) were highly homologous to carp Cyprinus carpio nuclear genes. The obtained data suggested that in the history of aquarium goldfish breeding occurred the stage of distant hybridization between goldfish and common carp. Subsequently, the nuclear genomes of some ornamental forms could be enriched by goldfish genes (a relatively recent form as Oriental twintail goldfish and Chinese Ranchu) or common carp genes (the simplest and most ancient forms like common goldfish) as a result of multidirectional breeding and selection of aquarium goldfish various forms. -

Electrophoretic Studies of Diploid, Triploid, and Tetraploid Forms of the Japanese Silver Crucian Carp, Carassius Auratus Langsdorfii
Japan.J.Ichthyol. 魚 類 学 雑 誌 40(1):65-75,1993 40(1):65-75,1993 Electrophoretic Studies of Diploid, Triploid, and Tetraploid Forms of the Japanese Silver Crucian Carp, Carassius auratus langsdorfii Yasushi Shimizu,1,2 Takashi Oshiro1 and Mitsuru Sakaizumi2,31 Department of Aquatic Biosciences, Tokyo University of Fisheries, 4-5-7 Konan, Minato-ku, Tokyo 108, Japan 2Department of Experimental Animal Science, The Tokyo Metropolitan Institute of Medical Science, 3-18-22 Honkomagome, Bunkyo-ku, Tokyo 113, Japan 3Present address: Department of Biology, College of General Education, Niigata University, 2-8050 Ikarashi, Niigata 950-21, Japan (Received October 29, 1992; in revisedform February 8, 1993; accepted March 22, 1993) Abstract The diploid-polyploid complex in the Japanese silver crucian carp (ginbuna), Carassius auratus langsdorfii, includes a diploid bisexual form, a triploid gynogenetic form and a tetraploid gynogenetic form. However, the origin of the polyploids remains to be clarified. Flow cytometric and electrophoretic analyses of Japanese crucian carp demonstrated that all the individuals with a two-banded pattern for an AMY-2 gene were polyploid, and 90% of the polyploids gave such a two-banded pattern. One of the two bands was not found in diploid subspecies in Japan but was present in both Korean crucian carp and goldfish. The diploid subspecies, kinbuna (C.auratus subsp.), which is distributed along the Pacific coast of eastern Japan, generated a diagnostic band of phosphoglucomutase. This band was also found in the polyploid "ginbuna" from the same region, but was observed in neither triploids nor diploids from other regions. -

Flood Loss Model Model
GIROJ FloodGIROJ Loss Flood Loss Model Model General Insurance Rating Organization of Japan 2 Overview of Our Flood Loss Model GIROJ flood loss model includes three sub-models. Floods Modelling Estimate the loss using a flood simulation for calculating Riverine flooding*1 flooded areas and flood levels Less frequent (River Flood Engineering Model) and large- scale disasters Estimate the loss using a storm surge flood simulation for Storm surge*2 calculating flooded areas and flood levels (Storm Surge Flood Engineering Model) Estimate the loss using a statistical method for estimating the Ordinarily Other precipitation probability distribution of the number of affected buildings and occurring disasters related events loss ratio (Statistical Flood Model) *1 Floods that occur when water overflows a river bank or a river bank is breached. *2 Floods that occur when water overflows a bank or a bank is breached due to an approaching typhoon or large low-pressure system and a resulting rise in sea level in coastal region. 3 Overview of River Flood Engineering Model 1. Estimate Flooded Areas and Flood Levels Set rainfall data Flood simulation Calculate flooded areas and flood levels 2. Estimate Losses Calculate the loss ratio for each district per town Estimate losses 4 River Flood Engineering Model: Estimate targets Estimate targets are 109 Class A rivers. 【Hokkaido region】 Teshio River, Shokotsu River, Yubetsu River, Tokoro River, 【Hokuriku region】 Abashiri River, Rumoi River, Arakawa River, Agano River, Ishikari River, Shiribetsu River, Shinano -

Keys to the Flesh Flies of Japan, with the Description of a New Genus And
〔Med. Entomol. Zool. Vol. 66 No. 4 p. 167‒200 2015〕 167 reference DOI: 10.7601/mez.66.167 Keys to the esh ies of Japan, with the description of a new genus and species from Honshu (Diptera: Sarcophagidae) Hiromu Kurahashi*, 1) and Susumu Kakinuma2) * Corresponding author: [email protected] 1) Department of Medical Entomology, National Institute of Infectious Diseases, Toyama 1‒23‒1, Shinjuku-ku, Tokyo 162‒8640 Japan 2) IDD Yamaguchi Lab., Aobadai 11‒22, Yamaguchi-shi, Yamaguchi 753‒0012 Japan (Received: 9 June 2015; Accepted: 2 October 2015) Abstract: A new genus and species of the Japanese Sarcophagidae, Papesarcophaga kisarazuensis gen. & sp. nov. is described and illustrated from Honshu, Japan. Practical keys to the Japanese 43 genera and 122 species are provided including this new species. A check list and data of specimens examined are also provided. Key words: Diptera, flesh flies, new species, new genus, Sarcophagidae, Japan INTRODUCTION The collection of Sarcophagidae made by the first author was studied during the course of the taxonomical studies on the calypterate muscoid flies from Japan since 1970 (Kurahashi, 1970). This was a revision of the subfamily Miltogramatinae dealing with seven genera and 14 species. Before this, Takano (1950) recorded seven genera and nine species of Japanese Sarcophagidae. Many investigation on the Japanese flesh flies made by Drs. K. Hori, R. Kano and S. Shinonaga beside the present authors. The results of these authors were published in the part of Sacophagidae, Fauna Japanica (Insecta: Diptera) and treated 23 genera and 65 species of the subfamilies of Sarcophaginae and Agriinae (=Paramacronychiinae), but the subfamily Miltogrammatinae was not included (Kano et al., 1967). -

PICES-2012 Program and Abstracts
PICES-2012 Program & Abstracts Image: “The Itsukushima Shrine from the sea”, November 25, 2010, watercolour, by Satoshi Arima, retiree of the National Research Institute of Fisheries and Environment of Inland Sea, Hiroshima, Japan. Permission to reproduce this artwork was kindly granted by Mr. Arima. Prepared and published by: North Pacific Marine Science Organization PICES Secretariat P.O. Box 6000 Sidney, BC PICES-2012 V8L 4B2 Canada Tel: 1-250-363-6366 Fax: 1-250-363-6827 Program and Abstracts E-mail: [email protected] Website: www.pices.int October 12-21, 2012 Hiroshima, Japan PICES-2012 Effects of natural and anthropogenic stressors in the North Pacific ecosystems: Scientific challenges and possible solutions North Pacific Marine Science Organization October 12-21, 2012 Hiroshima, Japan Table of Contents Notes for Guidance � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �vi International Conference Center Floor Plan � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � vii Meeting Timetable � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �viii Map of the International Conference Center Area � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � xii Keynote Lecture � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 1 Schedules and Abstracts S1: Science Board Symposium Effects of natural and anthropogenic stressors in -

PHYLOGENY and ZOOGEOGRAPHY of the SUPERFAMILY COBITOIDEA (CYPRINOIDEI, Title CYPRINIFORMES)
PHYLOGENY AND ZOOGEOGRAPHY OF THE SUPERFAMILY COBITOIDEA (CYPRINOIDEI, Title CYPRINIFORMES) Author(s) SAWADA, Yukio Citation MEMOIRS OF THE FACULTY OF FISHERIES HOKKAIDO UNIVERSITY, 28(2), 65-223 Issue Date 1982-03 Doc URL http://hdl.handle.net/2115/21871 Type bulletin (article) File Information 28(2)_P65-223.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP PHYLOGENY AND ZOOGEOGRAPHY OF THE SUPERFAMILY COBITOIDEA (CYPRINOIDEI, CYPRINIFORMES) By Yukio SAWADA Laboratory of Marine Zoology, Faculty of Fisheries, Bokkaido University Contents page I. Introduction .......................................................... 65 II. Materials and Methods ............... • • . • . • . • • . • . 67 m. Acknowledgements...................................................... 70 IV. Methodology ....................................•....•.........•••.... 71 1. Systematic methodology . • • . • • . • • • . 71 1) The determinlttion of polarity in the morphocline . • . 72 2) The elimination of convergence and parallelism from phylogeny ........ 76 2. Zoogeographical methodology . 76 V. Comparative Osteology and Discussion 1. Cranium.............................................................. 78 2. Mandibular arch ...................................................... 101 3. Hyoid arch .......................................................... 108 4. Branchial apparatus ...................................•..••......••.. 113 5. Suspensorium.......................................................... 120 6. Pectoral -

Direct Cytotoxic Activity of CD8+ T Cells Against Ichthyophthirius Multifiliis
426 Abstracts / Fish & Shellfish Immunology 91 (2019) 421e472 importance for stress effects on the immune response in teleosts. Indi- # Corresponding author. vidual aspects of the interference of stress hormones (mainly cortisol) with E-mail address: [email protected] (J. Zou). immune processes have already been reported in some bony fish. Although less studied, the catecholamines adrenaline and noradrenaline have also shown to modulate the immune response of teleost leukocytes via a and b adrenergic receptors. This study aims to expand the actual knowledge on stress-induced immune modulation, in order to evaluate O-014. the effects of stress on the immune system of maraena whitefish (Cor- Direct cytotoxic activity of CD8þ T cells against Ichthyophthirius egonus maraena). This salmonid fish is highly sensitive to stress compared multifiliis in ginbuna crucian carp, Carassius auratus langsdorfii to other salmonid species long adapted to aquaculture. To this end, a large set of specific primers was designed for reverse-transcription quantitative Masaki Sukeda, Koumei Shiota, Takahiro Nagasawa, Miki Nakao, real-time PCR (RT-qPCR) analyses. The primer panel included cell-specific Tomonori Somamoto#. marker genes characterizing the distinct cell populations in the head kidney of C. maraena, which had been sorted using flow cytometry. In Laboratory of Marine Biochemistry, Department of Bioscience and addition, we analysed the expression of catecholamine and cortisol re- Biotechnology, Graduate School of Bioresource and Bioenvironmental ceptors in each population, in order to define the repertoire of stress- Sciences, Kyushu University, Fukuoka, Japan related modulators present in the cells. In the next step, we performed a series of in vitro stimulations of head kidney leukocytes to study the Abstract expression of genes involved in immune activation and acute phase A line of studies has shown that several humoral immune factors including together with catecholamine and cortisol receptors. -

In Vitro Characteristics of Cyprinid Herpesvirus 2: Effect of Kidney Extract Supplementation on Growth
Vol. 115: 223–232, 2015 DISEASES OF AQUATIC ORGANISMS Published August 20 doi: 10.3354/dao02885 Dis Aquat Org In vitro characteristics of cyprinid herpesvirus 2: effect of kidney extract supplementation on growth Tomoya Shibata1, Azusa Nanjo1, Masato Saito1, Keisuke Yoshii1, Takafumi Ito2, Teruyuki Nakanishi3, Takashi Sakamoto1, Motohiko Sano1,* 1Faculty of Marine Science, Tokyo University of Marine Science and Technology, Minato, Tokyo 108-8477, Japan 2Aquatic Animal Health Division, National Research Institute of Aquaculture, Fisheries Research Agency, Tamaki, Mie 519-0423, Japan 3Department of Veterinary Medicine, Nihon University, Fujisawa, Kanagawa 252-0880, Japan ABSTRACT: Herpesviral hematopoietic necrosis caused by goldfish hematopoietic necrosis virus (now identified as cyprinid herpesvirus 2, CyHV-2) has contributed to economic losses in goldfish Carassius auratus culture and is becoming a major obstacle in Prussian carp C. gibelio aquacul- ture in China. Several reports have described difficulties in culturing the virus, with the total loss of infectivity within several passages in cell culture. We succeeded in propagating CyHV-2 with a high infectious titer in a RyuF-2 cell line newly derived from the fin of the Ryukin goldfish variety using culture medium supplemented with 0.2% healthy goldfish kidney extract. The addition of kidney extract to the medium enabled rapid virus growth, resulting in the completion of cyto- pathic effect (CPE) within 4 to 6 d at 25°C. The extract also enabled reproducible virus culture 5−6 −1 with a titer of 10 TCID50 ml . The virus cultured using this protocol showed pathogenicity in goldfish after intraperitoneal injection. The virus grew in RyuF-2 cells at 15, 20, 25, 30, and 32°C but not at 34°C or higher. -

Japan: Tokai Heavy Rain (September 2000)
WORLD METEOROLOGICAL ORGANIZATION THE ASSOCIATED PROGRAMME ON FLOOD MANAGEMENT INTEGRATED FLOOD MANAGEMENT CASE STUDY1 JAPAN: TOKAI HEAVY RAIN (SEPTEMBER 2000) January 2004 Edited by TECHNICAL SUPPORT UNIT Note: Opinions expressed in the case study are those of author(s) and do not necessarily reflect those of the WMO/GWP Associated Programme on Flood Management (APFM). Designations employed and presentations of material in the case study do not imply the expression of any opinion whatever on the part of the Technical Support Unit (TSU), APFM concerning the legal status of any country, territory, city or area of its authorities, or concerning the delimitation of its frontiers or boundaries. WMO/GWP Associated Programme on Flood Management JAPAN: TOKAI HEAVY RAIN (SEPTEMBER 2000) Ministry of Land, Infrastructure and Transport, Japan 1. Place 1.1 Location Positions in the flood inundation area caused by the Tokai heavy rain: Nagoya City, Aichi Prefecture is located at 35° – 35° 15’ north latitude, 136° 45’ - 137° east longitude. The studied area is Shonai and Shin river basin- hereinafter referred to as the Shonai river system. It locates about the center of Japan including Nagoya city area, 5th largest city in Japan with the population about 3millions. Therefore, two rivers flow through densely populated area and into the Pacific Ocean and are typical city-type rivers in Japan. Shin Riv. Border of basin Shonai Riv. Flooding area Point of breach ●Peak flow rate in major points on Sept. 12 (app. m3/s) ← Nagoya City, ← ← ino ino Aichi Prefecture j Ku ← 1,100 Shin Riv. ← 720 ← → ← ima Detention j Basin Shinkawa Araizeki Shidami Biwa (Fixed dam) Shin Riv.