Review Telaprevir: Pharmacokinetics and Drug Interactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Telaprevir (Incivek)
© Hepatitis C Online PDF created September 25, 2021, 4:19 pm Telaprevir (Incivek) Discontinued. This treatment has been discontinued. Table of Contents Telaprevir Incivek Summary Drug Summary Adverse Effects Class and Mechanism Manufacturer for United States FDA Status Indications Dosing Clinical Use Cost and Medication Access Resistance Key Drug Interactions Full Prescribing Information Figures Drug Summary Although telaprevir was a promising direct-acting antiviral agent that had impact in the hepatitis C treatment field during 2011 to 2013, it was subsequently replaced by newer direct-acting antiviral agents that were more effective, better tolerated, and more convenient. Based on the dwindling role of telaprevir after newer direct-acting antiviral agents were approved, Vertex pharmaceuticals discontinued the sales and distribution of telaprevir in the United States in October 2014. Telaprevir does have some current importance since persons who previously failed a telaprevir-based regimen may have developed resistant associated variants, which could potentially impact subsequent therapy. Adverse Effects The most significant adverse effects reported in the main registration trials and in post-marketing experience were rash, anorectal complaints, and anemia. When comparing triple therapy of telaprevir, peginterferon, and ribavirin with dual therapy of peginterferon and ribavirin alone significant differences were noted with rash (56% versus 34%), anemia (36% versus 17%), and anorectal complaints that include anorectal discomfort, anal pruritus, and hemorrhoids (29% versus 7%). In most cases, the rash that develops is eczematous or maculopapular in character and mild to moderate in severity; the rash is typically manageable with good skin care and topical emollients or corticosteroids. In some instances, however, telaprevir has caused serious skin Page 1/5 rashes, including Steven's Johnson Syndrome (SJS), Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), and Toxic Epidermal Necrolysis (TEN). -

Telaprevir for HIV/Hepatitis C Virus–Coinfected Patients Failing
HIV/AIDS MAJOR ARTICLE Telaprevir for HIV/Hepatitis C Virus–Coinfected Patients Failing Treatment With Pegylated Interferon/Ribavirin (ANRS HC26 TelapreVIH): An Open-Label, Single-Arm, Phase 2 Trial Downloaded from https://academic.oup.com/cid/article/59/12/1768/2895305 by guest on 01 October 2021 Laurent Cotte,1 Joséphine Braun,2 Caroline Lascoux-Combe,3 Corine Vincent,2 Marc-Antoine Valantin,4 Philippe Sogni,5 Karine Lacombe,6 Didier Neau,7 Hugues Aumaitre,8 Dominique Batisse,9 Pierre de Truchis,10 Anne Gervais,11 Christian Michelet,12 Philippe Morlat,13 Daniel Vittecoq,14 Isabelle Rosa,15 Inga Bertucci,16 Stéphane Chevaliez,17 Jean-Pierre Aboulker,2 and Jean-Michel Molina3; for the French National Agency for Research on AIDS and Viral Hepatitis (ANRS) HC26 Study Groupa 1Hospices Civils de Lyon, Croix-Rousse Hospital, and INSERM U1052, 2INSERM SC10-US019, Villejuif, 3Assistance Publique–Hôpitaux de Paris (AP-HP), Saint-Louis Hospital, University of Paris VII Denis Diderot, and Sorbonne Paris-Cité, INSERM U941, 4AP-HP, Pitié-Salpêtrière Hospital, and UMR-S 943, INSERM, 5AP-HP, Cochin Hospital and Paris Descartes University, INSERM U-1016, 6AP-HP, Saint-Antoine Hospital, Sorbonne Universités, UPMC University Paris 06, UMR-S1136, 7Pellegrin University Hospital, Bordeaux, 8Saint-Jean Hospital, Perpignan, 9AP-HP, Georges Pompidou European Hospital, Paris, 10AP-HP, Raymond Poincaré Hospital, Garches, 11AP-HP, Bichat-Claude Bernard Hospital, Paris, 12Pontchaillou University Hospital, Rennes, 13Saint-André University Hospital, Bordeaux, 14AP-HP, Bicètre Hospital, Le Kremlin-Bicètre, 15Créteil Hospital, 16French National Agency for Research on AIDS and Viral Hepatitis, Paris, and 17AP-HP, Henri Mondor Hospital, Créteil, France (See the Editorial Commentary by Rockstroh on pages 1777–8.) Background. -

The Legally Binding Text Is the Original French Version TRANSPARENCY
The legally binding text is the original French version TRANSPARENCY COMMITTEE OPINION 14 December 2011 INCIVO 375 mg, film-coated tablet B/4 bottles of 42 tablets (CIP code: 217 378-5) B/1 bottle of 42 tablets (CIP code: 219 249-8) Applicant: JANSSEN-CILAG telaprevir ATC Code: J05AE11 (protease inhibitor) List I Hospital prescription restricted to gastroenterology, hepatology, internal medicine or infectiology specialists. Date of Marketing Authorisation (centralised European procedure): Box of 4 bottles: 19/09/2011 Box of 1 bottle: 13/10/2011 Reason for request: Inclusion on the list of medicines refundable by National Health Insurance and on the list of medicines approved for hospital use. Medical, Economic and Public Health Assessment Division 1/22 1. CHARACTERISTICS OF THE MEDICINAL PRODUCT 1.1. Active ingredient telaprevir 1.2. Background This is an NS3/4A protease inhibitor of the genotype 1 hepatitis C virus. 1.3. Indication “INCIVO, in combination with peginterferon alfa and ribavirin, is indicated for the treatment of genotype 1 chronic hepatitis C in adult patients with compensated liver disease (including cirrhosis): - who are treatment-naive; - who have previously been treated with interferon alfa (pegylated or non-pegylated) alone or in combination with ribavirin, including relapsers, partial responders and null responders (see sections 4.4 and 5.1 of the SPC).” 1.4. Dosage “Treatment with INCIVO must be initiated and monitored by a physician experienced in the management of chronic hepatitis C. INCIVO, 750 mg dose of INCIVO (two 375 mg film-coated tablets) should be taken orally every 8 hours with food (the total daily dose is 6 tablets (2,250 mg)). -

Caracterización Molecular Del Perfil De Resistencias Del Virus De La
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Programa de doctorado en Medicina Departamento de Medicina Facultad de Medicina Universidad Autónoma de Barcelona TESIS DOCTORAL Caracterización molecular del perfil de resistencias del virus de la hepatitis C después del fallo terapéutico a antivirales de acción directa mediante secuenciación masiva Tesis para optar al grado de doctor de Qian Chen Directores de la Tesis Dr. Josep Quer Sivila Dra. Celia Perales Viejo Dr. Josep Gregori i Font Laboratorio de Enfermedades Hepáticas - Hepatitis Víricas Vall d’Hebron Institut de Recerca (VHIR) Barcelona, 2018 ABREVIACIONES Abreviaciones ADN: Ácido desoxirribonucleico AK: Adenosina quinasa ALT: Alanina aminotransferasa ARN: Ácido ribonucleico ASV: Asunaprevir BOC: Boceprevir CCD: Charge Coupled Device CLDN1: Claudina-1 CHC: Carcinoma hepatocelular DAA: Antiviral de acción directa DC-SIGN: Dendritic cell-specific ICAM-3 grabbing non-integrin DCV: Daclatasvir DSV: Dasabuvir -
PASS Information
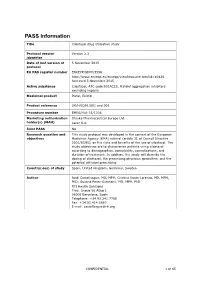
PASS Information Title Cilostazol drug utilisation study Protocol version Version 2.3 identifier Date of last version of 5 November 2015 protocol EU PAS register number ENCEPP/SDPP/3596 http://www.encepp.eu/encepp/viewResource.htm?id=10426 Accessed 5 November 2015 Active substance Cilostazol, ATC code B01AC23, Platelet aggregation inhibitors excluding heparin Medicinal product Pletal, Ekistol Product reference UK/H/0291/001 and 002 Procedure number EMEA/H/A-31/1306 Marketing authorisation Otsuka Pharmaceutical Europe Ltd. holder(s) (MAH) Lacer S.A. Joint PASS No Research question and This study protocol was developed in the context of the European objectives Medicines Agency (EMA) referral (article 31 of Council Directive 2001/83/EC) on the risks and benefits of the use of cilostazol. The study objectives are to characterise patients using cilostazol according to demographics, comorbidity, comedications, and duration of treatment. In addition, the study will describe the dosing of cilostazol, the prescribing physician specialties, and the potential off-label prescribing. Country(-ies) of study Spain, United Kingdom, Germany, Sweden Author Jordi Castellsague, MD, MPH; Cristina Varas-Lorenzo, MD, MPH, PhD; Susana Perez-Gutthann, MD, MPH, PhD RTI Health Solutions Trav. Gracia 56 Atico 1 08006 Barcelona, Spain Telephone: +34.93.241.7766 Fax: +34.93.414.2610 E-mail: [email protected] CONFIDENTIAL 1 of 65 Marketing Authorisation Holder(s) Marketing authorisation Otsuka Pharmaceutical Europe Ltd. holder(s) Gallions Wexham Springs Framewood Road Wexham SL3 6PJ, UK Lacer S.A. Sardenya 350 08025 Barcelona Spain MAH contact person Dr Marco Avila Regional Vice President, Medical Europe Otsuka Pharmaceutical Europe Ltd EU QPPV Dr Achint Kumar Otsuka Europe Development and Commercialisation Limited (OEDC) Head of Medical Dr Sanjay Kapoor Compliance-Europe Otsuka Pharmaceutical Europe Ltd. -

Medicinal Product No Longer Authorised
Assessment report INCIVO telaprevir Procedure No.: EMEA/H/C/002313/ Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. Medicinal product no longer authorised 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8613 E-mail [email protected] Website www.ema.europaeu An agency of the European Union © European Medicines Agency, 2011. Reproduction is authorised provided the source is acknowledged. 21 July 2011 EMA/CHMP/475470/2011 Committee for Medicinal Products for Human Use (CHMP) CHMP assessment report INCIVO International non-proprietary name: telaprevir authorised Procedure No. EMEA/H/C/002313 longer no product Medicinal 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8613 E-mail [email protected] Website www.ema.europaeu An agency of the European Union CHMP assessment report Page 2/109 Product information Name of the medicinal product: INCIVO Applicant: Janssen-Cilag International N.V. Turnhoutseweg 30 BE-2340 Beerse Belgium Active substance: telaprevir International Nonproprietary Name/Common Name: telaprevir Pharmaco-therapeutic group Protease inhibitors authorised (ATC Code): (J05AE) INCIVO, in combination with peginterferon alfa and ribavirin, is indicated for the treatment of genotype Therapeutic indication(s): 1 chronic hepatitis C in adult patients with compensated liver disease (including cirrhosis): - who are treatment-naïve; - who have previously been treated with interferonlonger alfa (pegylated or non-pegylated) alone or in combination with ribavirin, including relapsers, partial responders and null responders (see sections 4.4 and 5.1). -

PASS Information Title Cilostazol Drug Utilisation Study
PASS Information Title Cilostazol drug utilisation study Protocol version Version 2 identifier Date of last version of February 28, 2013 protocol EU PAS register number Registration is planned prior to study initiation once the protocol is final and has received regulatory endorsement by the Committee for Medicinal Products for Human Use (CHMP) Active substance Cilostazol, ATC code B01AC23, Platelet aggregation inhibitors excluding heparin Medicinal product Pletal, Ekistol Product reference UK/H/0291/001 and 002 Procedure number EMEA/H/A-31/1306 Marketing authorisation Otsuka Pharmaceutical Europe Ltd. holder(s) (MAH) Lacer S.A. Joint PASS No Research question and This study protocol was developed in the context of the European objectives Medicines Agency (EMA) referral (article 31 of Council Directive 2001/83/EC) on the risks and benefits of the use of cilostazol. The study objectives are to characterise patients using cilostazol according to demographics, comorbidity, comedications, and duration of treatment. In addition, the study will describe the dosing of cilostazol, the prescribing physician specialties, and the potential off-label prescribing. Country(-ies) of study Spain, United Kingdom, Germany, Sweden Author Jordi Castellsague, MD, MPH; Cristina Varas-Lorenzo, MD, MPH, PhD; Susana Perez-Gutthann, MD, MPH, PhD RTI Health Solutions Trav. Gracia 56 Atico 1 08006 Barcelona, Spain Telephone: +34.93.241.7766 Fax: +34.93.414.2610 E-mail: [email protected] CONFIDENTIAL 1 of 56 Marketing Authorisation Holder(s) Marketing authorisation -

Inhibitor Development Against P7 Channel in Hepatitis C Virus
molecules Article Inhibitor Development against p7 Channel in Hepatitis C Virus Shukun Wei 1,2,†, Xiaoyou Hu 2,3,†, Lingyu Du 1, Linlin Zhao 1,4, Hongjuan Xue 5, Chaolun Liu 6, James J. Chou 4, Jin Zhong 2,3,6, Yimin Tong 3,*, Shuqing Wang 7,* and Bo OuYang 1,2,* 1 State Key Laboratory of Molecular Biology, Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 333 Haike Road, Shanghai 201203, China; [email protected] (S.W.); [email protected] (L.D.); [email protected] (L.Z.) 2 University of Chinese Academy of Sciences, Beijing 100049, China; [email protected] (X.H.); [email protected] (J.Z.) 3 CAS Key Laboratory of Molecular Virology and Immunology, Institute Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai 200031, China 4 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA; [email protected] 5 National Facility for Protein Science in Shanghai, ZhangJiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China; [email protected] 6 ShanghaiTech University, Shanghai 201210, China; [email protected] 7 School of Pharmacy, Tianjin Medical University, Tianjin 300070, China * Correspondence: [email protected] (Y.T.); [email protected] (S.W.); [email protected] (B.O.) † This authors contributed equally. Abstract: Hepatitis C Virus (HCV) is the key cause of chronic and severe liver diseases. The recent direct-acting antiviral agents have shown the clinical success on HCV-related diseases, but the rapid HCV mutations of the virus highlight the sustaining necessity to develop new drugs. -

Symmetrel (Amantadine)
An#virals Lecture 20 Biology W3310/4310 Virology Spring 2015 You can’t go back and you can’t stand s0ll. If the thunder don’t get you, then the lightning will. JERRY GARCIA The Wheel (lyrics by Robert Hunter) Vaccines can prevent viral disease • But they Have modest or no therapeuMc effect if an individual is already infected (excepMon?) • Our second arm of anMviral defense is anMvirals • Can stop infecMon once it Has started Despite 50 years of research, our arsenal of an#viral drugs remains dangerously small Only about 100 an0viral drugs are available on the US market Most against HIV, HCV, herpesviruses - Persistent infecons Example of approved Target Virus compound Attachment HIV Maraviroc Uncoating Influenza A Amantadine, rimantadine Acyclovir, valacyclovir, DNA polymerase Herpesviruses cidofovir Reverse transcriptase HIV Zidovudine, nevirapine HIV Saquinavir, ritonavir Viral protease HCV Telaprevir, boceprevir Neuraminidase Influenza A and B Zanamavir, oseltamivir Why are there so few anviral drugs? • Compounds interfering with virus growth can adversely affect the Host cell - Side effects are common (unacceptable) - Every step in viral life cycle engages host funcons • Some medically important viruses can’t be propagated, Have no animal model, or are dangerous - HBV, HPV, norovirus - Smallpox - Ebola, Lassa An unappreciated third reason may be the most important • A compound must block virus replicaMon completely! It must be potent • Many standard pHarmaceuMcals can be effecMve if enZyme acMvity is parMally blocked • ParMal inHibiMon -

Usage of Antivirals and the Occurrence of Antiviral Resistance in Norway 2018
REPORT 2019 Usage of Antivirals and the Occurrence of Antiviral Resistance in Norway 2018 RAVN Resistensovervåking av virus i Norge Resistance against Antivirals in Norway Usage of Antivirals and the Occurrence of Antiviral resistance in Norway 2018 RAVN Resistensovervåkning av virus i Norge Resistance against antivirals in Norway 2 Published by the Norwegian Institute of Public Health Division of Infection Control and Environmental Health Department for Infectious Disease registries October 2019 Title: Usage of Antivirals and the Occurrence of Antiviral Resistance in Norway 2018. RAVN Ordering: The report can be downloaded as a pdf at www.fhi.no Graphic design cover: Fete Typer ISBN nr: 978-82-8406-032-3 Emneord (MeSH): Antiviral resistance Any usage of data from this report should include a specific reference to RAVN. Suggested citation: RAVN. Usage of Antivirals and the Occurrence of Antiviral Resistance in Norway 2018. Norwegian Institute of Public Health, Oslo 2019 Resistance against antivirals in Norway • Norwegian Institute of Public Health 3 Table of contents Introduction _________________________________________________________________________ 4 Contributors and participants __________________________________________________________ 5 Sammendrag ________________________________________________________________________ 6 Summary ___________________________________________________________________________ 8 1 Antivirals and development of drug resistance ______________________________________ 10 2 The usage of antivirals in Norway -

Estonian Statistics on Medicines 2013 1/44
Estonian Statistics on Medicines 2013 DDD/1000/ ATC code ATC group / INN (rout of admin.) Quantity sold Unit DDD Unit day A ALIMENTARY TRACT AND METABOLISM 146,8152 A01 STOMATOLOGICAL PREPARATIONS 0,0760 A01A STOMATOLOGICAL PREPARATIONS 0,0760 A01AB Antiinfectives and antiseptics for local oral treatment 0,0760 A01AB09 Miconazole(O) 7139,2 g 0,2 g 0,0760 A01AB12 Hexetidine(O) 1541120 ml A01AB81 Neomycin+Benzocaine(C) 23900 pieces A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+Thymol(dental) 2639 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+Cetylpyridinium chloride(gingival) 179340 g A01AD81 Lidocaine+Cetrimide(O) 23565 g A01AD82 Choline salicylate(O) 824240 pieces A01AD83 Lidocaine+Chamomille extract(O) 317140 g A01AD86 Lidocaine+Eugenol(gingival) 1128 g A02 DRUGS FOR ACID RELATED DISORDERS 35,6598 A02A ANTACIDS 0,9596 Combinations and complexes of aluminium, calcium and A02AD 0,9596 magnesium compounds A02AD81 Aluminium hydroxide+Magnesium hydroxide(O) 591680 pieces 10 pieces 0,1261 A02AD81 Aluminium hydroxide+Magnesium hydroxide(O) 1998558 ml 50 ml 0,0852 A02AD82 Aluminium aminoacetate+Magnesium oxide(O) 463540 pieces 10 pieces 0,0988 A02AD83 Calcium carbonate+Magnesium carbonate(O) 3049560 pieces 10 pieces 0,6497 A02AF Antacids with antiflatulents Aluminium hydroxide+Magnesium A02AF80 1000790 ml hydroxide+Simeticone(O) DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 34,7001 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 3,5364 A02BA02 Ranitidine(O) 494352,3 g 0,3 g 3,5106 A02BA02 Ranitidine(P) -

New Agents for the Treatment of Hepatitis C Virus – Focus on Telaprevir
Virus Adaptation and Treatment Dovepress open access to scientific and medical research Open Access Full Text Article REVIEW New agents for the treatment of hepatitis C virus – focus on telaprevir AJ Thompson1–3 Abstract: Antiviral therapy for hepatitis C virus (HCV) is rapidly evolving with the advent of K Patel3 direct-acting antiviral agents. Telaprevir is a first-generation linear ketoamide inhibitor of HCV NS3 protease. Approved in 2011 as standard-of-care for the treatment of patients chronically 1Department of Gastroenterology, St Vincent’s Hospital, University of infected with HCV genotype 1, telaprevir represents a major therapeutic advance. Used in com- Melbourne, 2Victorian Infectious bination with PEGylated interferon-alfa and ribavirin, telaprevir-based regimens cured . 75% Diseases Reference Laboratory, North Melbourne, Victoria, Australia; of treatment-naïve patients in the Phase III registration studies. Telaprevir is also effective for 3Duke Clinical Research Institute patients who have previously failed interferon-based therapy. Telaprevir presents a number of new and Department of Gastroenterology, challenges for clinicians, including a more demanding dosing schedule, telaprevir-specific adverse Duke University Medical Center, Durham, NC, USA events, potential for drug–drug interactions, and selection of drug-resistant HCV variants. Keywords: telaprevir, HCV, NS3, protease inhibitor, resistance Introduction There are up to 170 million individuals chronically infected with hepatitis C virus (HCV) worldwide, all of whom are at risk of the long-term complications of cirrhosis, liver failure, and hepatocellular carcinoma.1 Chronic HCV infection is currently the leading indication for liver transplantation in the Western world. Furthermore, without intervention, the burden of disease secondary to HCV infection is projected to increase over the coming decades as the HCV population ages.