Structures of Bacterial Rnd Transporters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unintentional Genomic Changes Endow Cupriavidus Metallidurans with an Augmented Heavy-Metal Resistance
G C A T T A C G G C A T genes Article Unintentional Genomic Changes Endow Cupriavidus metallidurans with an Augmented Heavy-Metal Resistance Felipe A. Millacura 1, Paul J. Janssen 2, Pieter Monsieurs 2, Ann Janssen 2, Ann Provoost 2, Rob Van Houdt 2 and Luis A. Rojas 3,* 1 School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3JQ, UK; [email protected] 2 Interdisciplinary Biosciences, Belgian Nuclear Research Centre, SCK•CEN, 2400 Mol, Belgium; [email protected] (P.J.J.); [email protected] (P.M.); [email protected] (A.J); [email protected] (A.P.); [email protected] (R.V.H.) 3 Chemistry Department, Faculty of Sciences, Universidad Católica del Norte, UCN, Antofagasta 1240000, Chile * Correspondence: [email protected]; Tel.: +56-55-235-5629 Received: 6 October 2018; Accepted: 8 November 2018; Published: 13 November 2018 Abstract: For the past three decades, Cupriavidus metallidurans has been one of the major model organisms for bacterial tolerance to heavy metals. Its type strain CH34 contains at least 24 gene clusters distributed over four replicons, allowing for intricate and multilayered metal responses. To gain organic mercury resistance in CH34, broad-spectrum mer genes were introduced in a previous work via conjugation of the IncP-1β plasmid pTP6. However, we recently noted that this CH34-derived strain, MSR33, unexpectedly showed an increased resistance to other metals (i.e., Co2+, Ni2+, and Cd2+). To thoroughly investigate this phenomenon, we resequenced the entire genome of MSR33 and compared its DNA sequence and basal gene expression profile to those of its parental strain CH34. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Table 4. V. Cholerae Flexgene ORF Collection
Table 4. V. cholerae FLEXGene ORF collection Reference Clone protein PlasmID clone GenBank Locus tag Symbol accession identifier FLEX clone name accession Product name VC0001 NP_062585 VcCD00019918 FLH200476.01F DQ772770 hypothetical protein VC0002 mioC NP_062586 VcCD00019938 FLH200506.01F DQ772771 mioC protein VC0003 thdF NP_062587 VcCD00019958 FLH200531.01F DQ772772 thiophene and furan oxidation protein ThdF VC0004 yidC NP_062588 VcCD00019970 FLH200545.01F DQ772773 inner membrane protein, 60 kDa VC0005 NP_062589 VcCD00061243 FLH236482.01F DQ899316 conserved hypothetical protein VC0006 rnpA NP_062590 VcCD00025697 FLH214799.01F DQ772774 ribonuclease P protein component VC0007 rpmH NP_062591 VcCD00061229 FLH236450.01F DQ899317 ribosomal protein L34 VC0008 NP_062592 VcCD00019917 FLH200475.01F DQ772775 amino acid ABC transporter, ATP-binding protein VC0009 NP_062593 VcCD00019966 FLH200540.01F DQ772776 amino acid ABC transproter, permease protein VC0010 NP_062594 VcCD00019152 FLH199275.01F DQ772777 amino acid ABC transporter, periplasmic amino acid-binding portion VC0011 NP_062595 VcCD00019151 FLH199274.01F DQ772778 hypothetical protein VC0012 dnaA NP_062596 VcCD00017363 FLH174286.01F DQ772779 chromosomal DNA replication initiator DnaA VC0013 dnaN NP_062597 VcCD00017316 FLH174063.01F DQ772780 DNA polymerase III, beta chain VC0014 recF NP_062598 VcCD00019182 FLH199319.01F DQ772781 recF protein VC0015 gyrB NP_062599 VcCD00025458 FLH174642.01F DQ772782 DNA gyrase, subunit B VC0016 NP_229675 VcCD00019198 FLH199346.01F DQ772783 hypothetical protein -

Supplemental Material-PDF
List of supplementary tables and figures. Table S1 – Strains sequenced and assembly information (n=72) Table S2 – Genetic clusters obtained using BAPS Table S3 – Genes commonly found to be under positive selection in the three subspecies (n=14) Table S4 - Recombining genes in X. fastidiosa subsp. pauca strains 9a5c and U24D (Citrus) Table S5 – Recombination genes for X. fastidiosa 32 (Coffee) Table S6 - Genes found to be under selection in each subspecies Table S7 – Recombining genes for each X. fastidiosa subspecies Table S8 – Genes found in the accessory genome of each subspecies Figure S1 - Phylogeny of X. fastidiosa strains (n=72) and pan-genome visualization Figure S2 - Detection of temporal signal in the dataset Figure S3 - Rate comparison amongst subspecies. The boxplot (A) was constructed my merging the rate of evolution of each branch (internal and external) of the X. fastidiosa subsp. fastidiosa, pauca and multiplex clades together (obtained though rate-dating) as illustrated by the colored frames (B) Figure S4 - Comparison between clonalFrameML and FastGEAR. (A) ClonalFrameML output, (B) FastGEAR output, (C) comparison between the genes found to be recombining between the two algorithms that were mapped to the X. fastidiosa reference genome Table S1 – Strains sequenced and assembly information (n=72). Strain names in parentheses represent synonyms. Name N50 L50 Total length Country Host Subspecies Isolation date Accession CO33 176,135 6 2,681,926 Italy Coffea arabica ? 2015 SAMN04100274 MUL0034 2,642,186 1 2,666,577 USA Morus alba morus 2003 SAMN03081485 Mul-MD 134,146 6 2,520,555 USA Morus alba morus 2011 SAMN02630185 Ann-1 44,355 22 2,729,755 USA Nerium oleander sandyi SAMN03081476 Xf_ATCC_35879 500,648 2 2,522,328 USA Vitis vinifera fastidiosa 1987 SAMN02997312 DSM10026 89,916 11 2,431,652 USA Vitis vinifera fastidiosa 1987 SAMN05660380 EB92-1 44,355 22 2,729,755 USA Sambucus sp. -

B Number Gene Name Strand Orientation Protein Length Mrna
list list sample) short list predicted B number Operon ID Gene name assignment Protein length mRNA present mRNA intensity Gene description Protein detected - Strand orientation Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0001 thrL + 21 1344 P 1 0 0 0 0 thr operon leader peptide Metabolism of small molecules 1 b0002 thrA + 820 13624 P 39 P 18 P 18 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules 1 b0003 thrB + 310 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules 1 b0004 thrC + 428 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules 1 b0005 b0005 + 98 432 A 5 0 0 0 0 orf, hypothetical protein Open reading frames 2 b0006 yaaA - 258 1047 P 11 P 1 2 P 1 0 orf, hypothetical protein Open reading frames 3 b0007 yaaJ - 476 342 P 8 0 0 0 0 MP-GenProt-PHD inner membrane transport protein Miscellaneous 4 b0008 talB + 317 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules 5 b0009 mog + 195 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules 6 b0010 yaaH - 188 407 A 2 0 0 0 0 PHD orf, hypothetical protein Open reading frames 7 b0011 b0011 - 237 338 P 13 0 0 0 0 putative oxidoreductase Miscellaneous 8 b0012 htgA -

The RHO Family Gtpases: Mechanisms of Regulation and Signaling
cells Review The RHO Family GTPases: Mechanisms of Regulation and Signaling Niloufar Mosaddeghzadeh and Mohammad Reza Ahmadian * Institute of Biochemistry and Molecular Biology II, Medical Faculty of the Heinrich Heine University, Universitätsstrasse 1, Building 22.03.05, 40225 Düsseldorf, Germany; [email protected] * Correspondence: [email protected] Abstract: Much progress has been made toward deciphering RHO GTPase functions, and many studies have convincingly demonstrated that altered signal transduction through RHO GTPases is a recurring theme in the progression of human malignancies. It seems that 20 canonical RHO GTPases are likely regulated by three GDIs, 85 GEFs, and 66 GAPs, and eventually interact with >70 downstream effectors. A recurring theme is the challenge in understanding the molecular determinants of the specificity of these four classes of interacting proteins that, irrespective of their functions, bind to common sites on the surface of RHO GTPases. Identified and structurally verified hotspots as functional determinants specific to RHO GTPase regulation by GDIs, GEFs, and GAPs as well as signaling through effectors are presented, and challenges and future perspectives are discussed. Keywords: CDC42; effectors; RAC1; RHOA; RHOGAP; RHOGDI; RHOGEF; RHO signaling 1. Introduction Citation: Mosaddeghzadeh, N.; The RHO (RAS homolog) family is an integral part of the RAS superfamily of guanine Ahmadian, M.R. The RHO Family nucleotide-binding proteins. RHO family proteins are crucial for several reasons: (i) ap- GTPases: Mechanisms of Regulation proximately 1% of the human genome encodes proteins that either regulate or are regulated and Signaling. Cells 2021, 10, 1831. by direct interaction with RHO proteins; (ii) they control almost all fundamental cellular https://doi.org/10.3390/cells10071831 processes in eukaryotes including morphogenesis, polarity, movement, cell division, gene expression, and cytoskeleton reorganization [1]; and (iii) they are associated with a series Academic Editor: Bor Luen Tang of human diseases (Figure1)[2]. -

Novel Genes Required for Surface-Associated Motility In
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.992537; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Novel genes required for surface-associated motility in 2 Acinetobacter baumannii 3 4 5 Ulrike Blaschke a,b,*, Evelyn Skiebe a and Gottfried Wilharm a,c,* 6 7 a Robert Koch-Institute, Project group P2, Burgstr. 37, D-38855 Wernigerode, 8 Germany 9 * Corresponding addresses: [email protected]; [email protected] 10 b ORCID: 0000-0002-3496-5447 11 c ORCID: 0000-0002-1771-6799 12 13 14 Keywords 15 Acinetobacter baumannii, surface-associated motility, transposon mutant library, 16 pellicle biofilm, antibiotic resistance, Galleria mellonella 17 18 Funding 19 This project was funded by the Deutsche Forschungsgemeinschaft (DFG) within FOR 20 2251 (WI 3272/3-1 and WI 3272/3-2). 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.992537; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 21 Abstract 22 Acinetobacter baumannii is an opportunistic and increasingly multi-drug resistant 23 human pathogen rated as a critical priority 1 pathogen for the development of new 24 antibiotics by the WHO in 2017. -

Polyamines Mitigate Antibiotic Inhibition of A.Actinomycetemcomitans Growth
Polyamines Mitigate Antibiotic Inhibition of A.actinomycetemcomitans Growth THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Allan Wattimena Graduate Program in Dentistry The Ohio State University 2017 Master's Examination Committee: Dr John Walters, Advisor Dr Purnima Kumar Dr Sara Palmer Dr Shareef Dabdoub Copyright by Allan Wattimena 2017 Abstract Polyamines are ubiquitous polycationic molecules that are present in all prokaryotic and eukaryotic cells. They are the breakdown products of amino acids and are important modulators of cell growth, stress and cell proliferation. Polyamines are present in higher concentrations in the periodontal pocket and may affect antibiotic resistance of bacterial biofilms. The effect of polyamines was investigated with amoxicillin (AMX), azithromycin (AZM) and doxycycline (DOX) on the growth of Aggregatibacter actinomycetemcomitans (A.a.) Y4 strain. Bacteria were grown in brain heart infusion broth under the following conditions: 1) A.a. only, 2) A.a. + antibiotic, 3) A.a. + antibiotic + polyamine mix (1.4mM putrescine, 0.4mM spermidine, 0.4mM spermine). Growth curve analysis, MIC determination and metatranscriptomic analysis were carried out. The presence of exogenous polyamines produced a small, but significant increase in growth of A.a. Polyamines mitigated the inhibitory effect of AMX, AZM and DOX on A.a. growth. Metatranscriptomic analysis revealed differing transcriptomic profiles when comparing AMX and AZM in the presence of polyamines. Polyamines produced a transient mitigation of AMX inhibition, but did not have a significant effect on gene transcription. Many gene transcription changes were seen when polyamines were in the presence of AZM. -
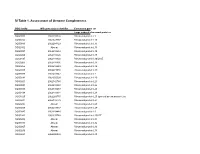
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Structure–Function Relationships of Glycoprotein Hormones and Their Subunits’ Ancestors
REVIEW ARTICLE published: 26 February 2015 doi: 10.3389/fendo.2015.00026 Structure–function relationships of glycoprotein hormones and their subunits’ ancestors Claire Cahoreau, Danièle Klett andYves Combarnous* Physiologie de la Reproduction et des Comportements (PRC), Centre National de la Recherche Scientifique, INRA, Nouzilly, France Edited by: Glycoprotein hormones (GPHs) are the most complex molecules with hormonal activity. Hubert Vaudry, University of Rouen, They exist only in vertebrates but the genes encoding their subunits’ ancestors are found France in most vertebrate and invertebrate species although their roles are still unknown. In the Reviewed by: Francisco Gaytán, University of present report, we review the available structural and functional data concerning GPHs and Cordoba, Spain their subunits’ ancestors. Bruno Querat, Université Keywords: glycoprotein hormones, luteinizing hormone, follicle-stimulating hormone, thyroid-stimulating Paris-Diderot, France hormone, evolution, molecular, structure–activity relationship *Correspondence: Yves Combarnous, Physiologie de la Reproduction et des Comportements (PRC), Centre National de la Recherche Scientifique, INRA, Nouzilly 37380, France e-mail: yves.combarnous@ tours.inra.fr INTRODUCTION In the present paper, we will consider the structure–function Glycoprotein hormones (GPHs) are the most complex mole- relationships of GPHs and of their receptors (GPHRs) to better cules with hormonal activity. They include three pituitary hor- understand their interactions and the subsequent steps in their mones, the gonadotropins follicle-stimulating hormone (FSH; target cells stimulation. follitropin) and luteinizing hormone (LH; lutropin) as well as thyroid-stimulating hormone (TSH; thyrotropin) (1). Only in pri- STRUCTURE OF GLYCOPROTEIN HORMONES AND THEIR mates (2) and equidaes (3), a chorionic gonadotropin (CG) is also ANCESTORS secreted by the placenta.