Lipid and Protein Organizations in Model Membrane Systems- Membrane Curvature, Lipid Structure, Domain Formation, and Membrane Binding Kinetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The PX Domain Protein Interaction Network in Yeast
The PX domain protein interaction network in yeast Zur Erlangung des akademischen Grades eines DOKTORS DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät für Chemie und Biowissenschaften der Universität Karlsruhe (TH) vorgelegte DISSERTATION von Dipl. Biol. Carolina S. Müller aus Buenos Aires Dekan: Prof. Dr. Manfred Kappes Referent: Dr. Nils Johnsson Korreferent: HD. Dr. Adam Bertl Tag der mündlichen Prüfung: 17.02.2005 I dedicate this work to my Parents and Alex TABLE OF CONTENTS Table of contents Introduction 1 Yeast as a model organism in proteome analysis 1 Protein-protein interactions 2 Protein Domains in Yeast 3 Classification of protein interaction domains 3 Phosphoinositides 5 Function 5 Structure 5 Biochemistry 6 Localization 7 Lipid Binding Domains 8 The PX domain 10 Function of PX domain containing proteins 10 PX domain structure and PI binding affinities 10 Yeast PX domain containing proteins 13 PX domain and protein-protein interactions 13 Lipid binding domains and protein-protein interactions 14 The PX-only proteins Grd19p and Ypt35p and their phenotypes 15 Aim of my PhD work 16 Project outline 16 Searching for interacting partners 16 Confirmation of obtained interactions via a 16 second independent method Mapping the interacting region 16 The Two-Hybrid System 17 Definition 17 Basic Principle of the classical Yeast-Two Hybrid System 17 Peptide Synthesis 18 SPOT synthesis technique 18 Analysis of protein- peptide contact sites based on SPOT synthesis 19 TABLE OF CONTENTS Experimental procedures 21 Yeast two-hybrid assay -

Chemical Agent and Antibodies B-Raf Inhibitor RAF265
Supplemental Materials and Methods: Chemical agent and antibodies B-Raf inhibitor RAF265 [5-(2-(5-(trifluromethyl)-1H-imidazol-2-yl)pyridin-4-yloxy)-N-(4-trifluoromethyl)phenyl-1-methyl-1H-benzp{D, }imidazol-2- amine] was kindly provided by Novartis Pharma AG and dissolved in solvent ethanol:propylene glycol:2.5% tween-80 (percentage 6:23:71) for oral delivery to mice by gavage. Antibodies to phospho-ERK1/2 Thr202/Tyr204(4370), phosphoMEK1/2(2338 and 9121)), phospho-cyclin D1(3300), cyclin D1 (2978), PLK1 (4513) BIM (2933), BAX (2772), BCL2 (2876) were from Cell Signaling Technology. Additional antibodies for phospho-ERK1,2 detection for western blot were from Promega (V803A), and Santa Cruz (E-Y, SC7383). Total ERK antibody for western blot analysis was K-23 from Santa Cruz (SC-94). Ki67 antibody (ab833) was from ABCAM, Mcl1 antibody (559027) was from BD Biosciences, Factor VIII antibody was from Dako (A082), CD31 antibody was from Dianova, (DIA310), and Cot antibody was from Santa Cruz Biotechnology (sc-373677). For the cyclin D1 second antibody staining was with an Alexa Fluor 568 donkey anti-rabbit IgG (Invitrogen, A10042) (1:200 dilution). The pMEK1 fluorescence was developed using the Alexa Fluor 488 chicken anti-rabbit IgG second antibody (1:200 dilution).TUNEL staining kits were from Promega (G2350). Mouse Implant Studies: Biopsy tissues were delivered to research laboratory in ice-cold Dulbecco's Modified Eagle Medium (DMEM) buffer solution. As the tissue mass available from each biopsy was limited, we first passaged the biopsy tissue in Balb/c nu/Foxn1 athymic nude mice (6-8 weeks of age and weighing 22-25g, purchased from Harlan Sprague Dawley, USA) to increase the volume of tumor for further implantation. -

Spatial and Temporal Aspects of Signalling 6 1
r r r Cell Signalling Biology Michael J. Berridge Module 6 Spatial and Temporal Aspects of Signalling 6 1 Module 6 Spatial and Temporal Aspects of Signalling Synopsis The function and efficiency of cell signalling pathways are very dependent on their organization both in space and time. With regard to spatial organization, signalling components are highly organized with respect to their cellular location and how they transmit information from one region of the cell to another. This spatial organization of signalling pathways depends on the molecular interactions that occur between signalling components that use signal transduction domains to construct signalling pathways. Very often, the components responsible for information transfer mechanisms are held in place by being attached to scaffolding proteins to form macromolecular signalling complexes. Sometimes these macromolecular complexes can be organized further by being localized to specific regions of the cell, as found in lipid rafts and caveolae or in the T-tubule regions of skeletal and cardiac cells. Another feature of the spatial aspects concerns the local Another important temporal aspect is timing and signal and global aspects of signalling. The spatial organization of integration, which relates to the way in which functional signalling molecules mentioned above can lead to highly interactions between signalling pathways are determined localized signalling events, but when the signalling mo- by both the order and the timing of their presentations. lecules are more evenly distributed, signals can spread The organization of signalling systems in both time and more globally throughout the cell. In addition, signals space greatly enhances both their efficiency and versatility. -

Modular Phosphoinositide-Binding Domains – Their Role in Signalling and Membrane Trafficking Peter J
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector R882 Review Modular phosphoinositide-binding domains – their role in signalling and membrane trafficking Peter J. Cullen*, Gyles E. Cozier*, George Banting† and Harry Mellor† The membrane phospholipid phosphatidylinositol is the Introduction precursor of a family of lipid second-messengers, The membrane phospholipid phosphatidylinositol is the known as phosphoinositides, which differ in the precursor for a family of lipid second-messengers, known phosphorylation status of their inositol group. A major collectively as phosphoinositides, that differ solely in the advance in understanding phosphoinositide signalling phosphorylation status of their inositol head group has been the identification of a number of highly (Figure 1) [1,2]. Phosphoinositides are ideally suited to conserved modular protein domains whose function function as spatially restricted membrane second messen- appears to be to bind various phosphoinositides. Such gers because: the synthesis and turnover of phosphoinosi- ‘cut and paste’ modules are found in a diverse array of tides from the relatively abundant phosphatidylinositol multidomain proteins and recruit their host protein to membrane precursor can be rapid and highly concentrated specific regions in cells via interactions with within discrete membrane micro domains; the ratio of phosphoinositides. Here, with particular reference to phosphoinositide to binding partner can be relatively large, -
238 Parametric P- Value FDR Geom Mean of Intensities in Class 1 (Plate
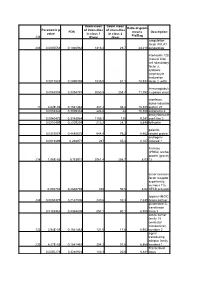
Geom mean Geom mean Ratio of geom Parametric p- of intensities of intensities FDR means Description value in class 1 in class 2 Pla/Bag 238 (Plate) (Bag) coagulation factor XIII, A1 206 0,0005558 0,1940962 1313,3 29,7 44,219 polypeptide interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation 0,0017883 0,2490398 1535,5 81,1 18,933 factor 2, p40) Immunoglobuli 0,0063804 0,3394787 3050,5 258,7 11,792 n epsilon chain interferon, alpha-inducible 11 4,62E-05 0,1541482 347,2 33,4 10,395 protein 27 0,0035346 0,3039332 226,8 22 10,309 astrotactin 2 deoxyribonucle 0,0040473 0,3148984 1155,1 125 9,241 ase I-like 3 0,0010495 0,2205294 213,2 24,1 8,846 follistatin galectin- 0,0315078 0,4885675 644,8 76,2 8,462 related protein androgen- 0,0013899 0,238971 287 35,4 8,107 induced 1 A kinase (PRKA) anchor protein (gravin) 236 1,06E-05 0,115911 2061,4 256,7 8,03 12 tumor necrosis factor receptor superfamily, member 11a, 0,002746 0,2846759 405 50,5 8,02 NFKB activator lipoma HMGIC 248 0,0008375 0,2147006 240,6 32,3 7,449 fusion partner glutathione S- transferase 0,0183462 0,4368809 557,1 80,1 6,955 theta 1 solute carrier family 18 (vesicular monoamine), 122 3,94E-05 0,1541482 121,5 17,6 6,903 member 2 signal transducing adaptor family 233 6,27E-05 0,1541482 354,3 51,6 6,866 member 1 Transcribed 0,0050276 0,3240923 140,3 20,6 6,811 locus aldehyde dehydrogenas e 5 family, member A1 (succinate- semialdehyde dehydrogenas 0,0294865 0,4819829 236,6 38,2 6,194 e) guanylate cyclase activator 1A 0,0074099 0,3504479 339,1 55 6,165 -

Deciphering the BAR Code of Membrane Modulators
Cell. Mol. Life Sci. DOI 10.1007/s00018-017-2478-0 Cellular and Molecular LifeSciences REVIEW Deciphering the BAR code of membrane modulators Ulrich Salzer1 · Julius Kostan2 · Kristina Djinović‑Carugo2,3 Received: 30 September 2016 / Revised: 25 January 2017 / Accepted: 27 January 2017 © The Author(s) 2017. This article is published with open access at Springerlink.com Abstract The BAR domain is the eponymous domain of Keywords N-BAR domain · F-BAR domain · I-BAR the “BAR-domain protein superfamily”, a large and diverse domain · lipid binding · Membrane remodelling · set of mostly multi-domain proteins that play eminent roles Membrane curvature at the membrane cytoskeleton interface. BAR domain homodimers are the functional units that peripherally asso- ciate with lipid membranes and are involved in membrane Introduction sculpting activities. Differences in their intrinsic curvatures and lipid-binding properties account for a large variety in Identification of sequence homology between the N-termi- membrane modulating properties. Membrane activities nal regions of the Bin, the amphiphysin, and the yeast Rvs of BAR domains are further modified and regulated by proteins led to the recognition of a novel protein domain intramolecular or inter-subunit domains, by intermolecu- which was named “BAR domain” as an acronym composed lar protein interactions, and by posttranslational modifica- of the first letters of these proteins [1]. This domain was tions. Rather than providing detailed cell biological infor- found in a large set of proteins which were classified as mation on single members of this superfamily, this review BAR domain proteins, later also termed N-BAR domain focuses on biochemical, biophysical, and structural aspects proteins, because several members of this protein family and on recent findings that paradigmatically promote our have an amphipathic helix at the N-terminus of the BAR understanding of processes driven and modulated by BAR domain [2]. -

Drosophila Epsin's Role in Notch Ligand Cells Requires Three
Developmental Biology 363 (2012) 399–412 Contents lists available at SciVerse ScienceDirect Developmental Biology journal homepage: www.elsevier.com/developmentalbiology Drosophila Epsin's role in Notch ligand cells requires three Epsin protein functions: The lipid binding function of the ENTH domain, a single Ubiquitin interaction motif, and a subset of the C-terminal protein binding modules Xuanhua Xie, Bomsoo Cho, Janice A. Fischer ⁎ Section of Molecular Cell and Developmental Biology, Institute for Cell and Molecular Biology, The University of Texas at Austin, 1 University Station A4800, Austin, Texas 78712, USA article info abstract Article history: Epsin is an endocytic protein that binds Clathrin, the plasma membrane, Ubiquitin, and also a variety of other Received for publication 17 September 2010 endocytic proteins through well-characterized motifs. Although Epsin is a general endocytic factor, genetic Revised 3 January 2012 analysis in Drosophila and mice revealed that Epsin is essential specifically for internalization of ubiquitinated Accepted 4 January 2012 transmembrane ligands of the Notch receptor, a process required for Notch activation. Epsin's mechanism of Available online 13 January 2012 function is complex and context-dependent. Consequently, how Epsin promotes ligand endocytosis and thus Notch signaling is unclear, as is why Notch signaling is uniquely dependent on Epsin. Here, by generating Keywords: Epsin Drosophila lines containing transgenes that express a variety of different Epsin deletion and substitution var- Notch signaling iants, we tested each of the five protein or lipid interaction modules for a role in Notch activation by each of Endocytosis the two ligands, Serrate and Delta. There are five main results of this work that impact present thinking about Drosophila the role of Epsin in ligand cells. -

International Phd Student Symposium and Career Fair for Life Sciences
16th Horizons in Molecular Biology 16TH HORIZONS IN MOLECULAR BIOLOGY International PhD Student Symposium and Career Fair for Life Sciences 9th-12th September 2019 Göttingen, Germany Bibliographical information held by the German National Library The German National Library has listed this book in the Deutsche National- bibliografie (German national bibliography); detailed bibliographic information is available online at http://dnb.d-nb.de. 1st edition - Göttingen: Cuvillier, 2019 © CUVILLIER VERLAG, Göttingen, Germany 2019 Nonnenstieg 8, 37075 Göttingen, Germany Telephone: +49 (0)551-54724-0 Telefax: +49 (0)551-54724-21 www.cuvillier.de ISBN 978-3-7369-9611-3 eISBN 978-3-7369-8611-4 This publication is printed on acid-free paper. © 2019 by Horizons in Molecular Biology PhD Student Organizing Committee Göttingen, Germany Booklet by Horizons Organizing team 2019, InDesign template provided by Sara Osman, Katharina Seitz, InDesign Layout: Claus-Peter Adam, MPI-BPC, Group Pictures: Irene Böttcher-Gajewski, MedienService, MPI-BPC All rights reserved. No part of this brochure may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means without the permission of the “Horizons in Molecular Biology” PhD Student Organizing Committee. This brochure is designed for informational purposes only. The PhD Student Organizing Committee cannot guarantee for accuracy and completeness and cannot be held liable for any direct, indirect or consequential damage arising from reliance upon this information. Acknowledgements It took almost twelve months to get us where we are now. Started with a big and extremely motivated team, we organized ourselves in smaller groups, each responsible for another field of organization. -

Inositol-Lipid Binding Motifs: Signal Integrators Through Protein-Lipid and Protein-Protein Interactions
Commentary 2093 Inositol-lipid binding motifs: signal integrators through protein-lipid and protein-protein interactions Tamas Balla Endocrinology and Reproduction Research Branch, NICHD, National Institutes of Health, Bethesda, MD 20892, USA Author for correspondence (e-mail: [email protected]) Accepted 16 March 2005 Journal of Cell Science 118, 2093-2104 Published by The Company of Biologists 2005 doi:10.1242/jcs.02387 Summary Inositol lipids have emerged as universal lipid regulators activities might influence one another through allosteric of protein signaling complexes in defined membrane mechanisms. Comparison of the structural features of these compartments. The number of protein modules that are domains not only reveals a high degree of conservation of known to recognise these membrane lipids is rapidly their lipid interaction sites but also highlights their increasing. Pleckstrin homology domains, FYVE domains, evolutionary link to protein modules known for protein- PX domains, ENTH domains, CALM domains, PDZ protein interactions. Protein-protein interactions involving domains, PTB domains and FERM domains are all lipid-binding domains could serve as the basis for inositide-recognition modules. The latest additions to this phosphoinositide-induced conformational regulation of list are members of the clathrin adaptor protein and target proteins at biological membranes. Therefore, these arrestin families. Initially, inositol lipids were believed modules function as crucially important signal integrators, to recruit signaling molecules -

EUROPEAN PATENT OFFICE, VIENNA Thousand Oaks, CA 91320 (US) SUB-OFFICE
Europäisches Patentamt *EP001033405A2* (19) European Patent Office Office européen des brevets (11) EP 1 033 405 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C12N 15/29, C12N 15/82, 06.09.2000 Bulletin 2000/36 C07K 14/415, C12Q 1/68, A01H 5/00 (21) Application number: 00301439.6 (22) Date of filing: 25.02.2000 (84) Designated Contracting States: • Brover, Vyacheslav AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU Calabasas, CA 91302 (US) MC NL PT SE • Chen, Xianfeng Designated Extension States: Los Angeles, CA 90025 (US) AL LT LV MK RO SI • Subramanian, Gopalakrishnan Moorpark, CA 93021 (US) (30) Priority: 25.02.1999 US 121825 P • Troukhan, Maxim E. 27.07.1999 US 145918 P South Pasadena, CA 91030 (US) 28.07.1999 US 145951 P • Zheng, Liansheng 02.08.1999 US 146388 P Creve Coeur, MO 63141 (US) 02.08.1999 US 146389 P • Dumas, J. 02.08.1999 US 146386 P , (US) 03.08.1999 US 147038 P 04.08.1999 US 147302 P (74) Representative: 04.08.1999 US 147204 P Bannerman, David Gardner et al More priorities on the following pages Withers & Rogers, Goldings House, (83) Declaration under Rule 28(4) EPC (expert 2 Hays Lane solution) London SE1 2HW (GB) (71) Applicant: Ceres Incorporated Remarks: Malibu, CA 90265 (US) THE COMPLETE DOCUMENT INCLUDING REFERENCE TABLES AND THE SEQUENCE (72) Inventors: LISTING IS AVAILABLE ON CD-ROM FROM THE • Alexandrov, Nickolai EUROPEAN PATENT OFFICE, VIENNA Thousand Oaks, CA 91320 (US) SUB-OFFICE. -

Multipurpose Cellular Lipids with Emerging Roles in Cell Death
Cell Death & Differentiation (2019) 26:781–793 https://doi.org/10.1038/s41418-018-0269-2 REVIEW ARTICLE Phosphoinositides: multipurpose cellular lipids with emerging roles in cell death 1 1 1 1 1 1 Thanh Kha Phan ● Scott A Williams ● Guneet K Bindra ● Fung T Lay ● Ivan K. H Poon ● Mark D Hulett Received: 5 September 2018 / Revised: 18 December 2018 / Accepted: 19 December 2018 / Published online: 11 February 2019 © ADMC Associazione Differenziamento e Morte Cellulare 2019 Abstract Phosphorylated phosphatidylinositol lipids, or phosphoinositides, critically regulate diverse cellular processes, including signalling transduction, cytoskeletal reorganisation, membrane dynamics and cellular trafficking. However, phosphoinosi- tides have been inadequately investigated in the context of cell death, where they are mainly regarded as signalling secondary messengers. However, recent studies have begun to highlight the importance of phosphoinositides in facilitating cell death execution. Here, we cover the latest phosphoinositide research with a particular focus on phosphoinositides in the mechanisms of cell death. This progress article also raises key questions regarding the poorly defined role of phosphoinositides, particularly during membrane-associated events in cell death such as apoptosis and secondary necrosis. fi 1234567890();,: 1234567890();,: The review then further discusses important future directions for the phosphoinositide eld, including therapeutically targeting phosphoinositides to modulate cell death. Facts peptide-induced necrosis, -

Interdependent Assembly of Specific Regulatory Lipids and Membrane
Published December 20, 2004 JCB: ARTICLE Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles Rutilio A. Fratti, Youngsoo Jun, Alexey J. Merz, Nathan Margolis, and William Wickner Department of Biochemistry, Dartmouth Medical School, Hanover, NH 03755 embrane microdomains are assembled by lipid ner. Regulatory lipids are also required for the vertex partitioning (e.g., rafts) or by protein–protein enrichment of SNAREs, Ypt7p, and HOPS. Conversely, M interactions (e.g., coated vesicles). During dock- SNAREs and actin regulate phosphatidylinositol 3-phos- ing, yeast vacuoles assemble “vertex” ring-shaped micro- phate vertex enrichment. Though the PX domain of the domains around the periphery of their apposed SNARE Vam7p has direct affinity for only 3-phosphoino- Downloaded from membranes. Vertices are selectively enriched in the Rab sitides, all the regulatory lipids which are needed for ver- GTPase Ypt7p, the homotypic fusion and vacuole protein tex assembly affect Vam7p association with vacuoles. sorting complex (HOPS)–VpsC Rab effector complex, Thus, the assembly of the vacuole vertex ring micro- SNAREs, and actin. Membrane fusion initiates at vertex domain arises from interdependent lipid and protein par- microdomains. We now find that the “regulatory lipids” titioning and binding rather than either lipid partitioning ergosterol, diacylglycerol and 3- and 4-phosphoinositides or protein interactions alone. jcb.rupress.org accumulate at vertices in a mutually interdependent man- Introduction on April 4, 2011 Membrane fusion is catalyzed by conserved factors (Jahn and membrane. The ring-shaped periphery of the boundary mem- Sudhof, 1999), including Rab family GTPases and their effectors brane is termed the “vertex”.