Accelerated Evolution of Oligodendrocytes in Human Brain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download (PDF)
ANALYTICAL SCIENCES NOVEMBER 2020, VOL. 36 1 2020 © The Japan Society for Analytical Chemistry Supporting Information Fig. S1 Detailed MS/MS data of myoglobin. 17 2 ANALYTICAL SCIENCES NOVEMBER 2020, VOL. 36 Table S1 : The protein names (antigens) identified by pH 2.0 solution in the eluted-fraction. These proteins were identified one or more out of six analyses. Accession Description P08908 5-hydroxytryptamine receptor 1A OS=Homo sapiens GN=HTR1A PE=1 SV=3 - [5HT1A_HUMAN] Q9NRR6 72 kDa inositol polyphosphate 5-phosphatase OS=Homo sapiens GN=INPP5E PE=1 SV=2 - [INP5E_HUMAN] P82987 ADAMTS-like protein 3 OS=Homo sapiens GN=ADAMTSL3 PE=2 SV=4 - [ATL3_HUMAN] Q9Y6K8 Adenylate kinase isoenzyme 5 OS=Homo sapiens GN=AK5 PE=1 SV=2 - [KAD5_HUMAN] P02763 Alpha-1-acid glycoprotein 1 OS=Homo sapiens GN=ORM1 PE=1 SV=1 - [A1AG1_HUMAN] P19652 Alpha-1-acid glycoprotein 2 OS=Homo sapiens GN=ORM2 PE=1 SV=2 - [A1AG2_HUMAN] P01011 Alpha-1-antichymotrypsin OS=Homo sapiens GN=SERPINA3 PE=1 SV=2 - [AACT_HUMAN] P01009 Alpha-1-antitrypsin OS=Homo sapiens GN=SERPINA1 PE=1 SV=3 - [A1AT_HUMAN] P04217 Alpha-1B-glycoprotein OS=Homo sapiens GN=A1BG PE=1 SV=4 - [A1BG_HUMAN] P08697 Alpha-2-antiplasmin OS=Homo sapiens GN=SERPINF2 PE=1 SV=3 - [A2AP_HUMAN] P02765 Alpha-2-HS-glycoprotein OS=Homo sapiens GN=AHSG PE=1 SV=1 - [FETUA_HUMAN] P01023 Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 - [A2MG_HUMAN] P01019 Angiotensinogen OS=Homo sapiens GN=AGT PE=1 SV=1 - [ANGT_HUMAN] Q9NQ90 Anoctamin-2 OS=Homo sapiens GN=ANO2 PE=1 SV=2 - [ANO2_HUMAN] P01008 Antithrombin-III -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Oligodendrocytes in Development, Myelin Generation and Beyond
cells Review Oligodendrocytes in Development, Myelin Generation and Beyond Sarah Kuhn y, Laura Gritti y, Daniel Crooks and Yvonne Dombrowski * Wellcome-Wolfson Institute for Experimental Medicine, Queen’s University Belfast, Belfast BT9 7BL, UK; [email protected] (S.K.); [email protected] (L.G.); [email protected] (D.C.) * Correspondence: [email protected]; Tel.: +0044-28-9097-6127 These authors contributed equally. y Received: 15 October 2019; Accepted: 7 November 2019; Published: 12 November 2019 Abstract: Oligodendrocytes are the myelinating cells of the central nervous system (CNS) that are generated from oligodendrocyte progenitor cells (OPC). OPC are distributed throughout the CNS and represent a pool of migratory and proliferative adult progenitor cells that can differentiate into oligodendrocytes. The central function of oligodendrocytes is to generate myelin, which is an extended membrane from the cell that wraps tightly around axons. Due to this energy consuming process and the associated high metabolic turnover oligodendrocytes are vulnerable to cytotoxic and excitotoxic factors. Oligodendrocyte pathology is therefore evident in a range of disorders including multiple sclerosis, schizophrenia and Alzheimer’s disease. Deceased oligodendrocytes can be replenished from the adult OPC pool and lost myelin can be regenerated during remyelination, which can prevent axonal degeneration and can restore function. Cell population studies have recently identified novel immunomodulatory functions of oligodendrocytes, the implications of which, e.g., for diseases with primary oligodendrocyte pathology, are not yet clear. Here, we review the journey of oligodendrocytes from the embryonic stage to their role in homeostasis and their fate in disease. We will also discuss the most common models used to study oligodendrocytes and describe newly discovered functions of oligodendrocytes. -

Neuregulin 1–Erbb2 Signaling Is Required for the Establishment of Radial Glia and Their Transformation Into Astrocytes in Cerebral Cortex
Neuregulin 1–erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex Ralf S. Schmid*, Barbara McGrath*, Bridget E. Berechid†, Becky Boyles*, Mark Marchionni‡, Nenad Sˇ estan†, and Eva S. Anton*§ *University of North Carolina Neuroscience Center and Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599; †Department of Neurobiology, Yale University School of Medicine, New Haven, CT 06510; and ‡CeNes Pharamceuticals, Inc., Norwood, MA 02062 Communicated by Pasko Rakic, Yale University School of Medicine, New Haven, CT, January 27, 2003 (received for review December 12, 2002) Radial glial cells and astrocytes function to support the construction mine whether NRG-1-mediated signaling is involved in radial and maintenance, respectively, of the cerebral cortex. However, the glial cell development and differentiation in the cerebral cortex. mechanisms that determine how radial glial cells are established, We show that NRG-1 signaling, involving erbB2, may act in maintained, and transformed into astrocytes in the cerebral cortex are concert with Notch signaling to exert a critical influence in the not well understood. Here, we show that neuregulin-1 (NRG-1) exerts establishment, maintenance, and appropriate transformation of a critical role in the establishment of radial glial cells. Radial glial cell radial glial cells in cerebral cortex. generation is significantly impaired in NRG mutants, and this defect can be rescued by exogenous NRG-1. Down-regulation of expression Materials and Methods and activity of erbB2, a member of the NRG-1 receptor complex, leads Clonal Analysis to Study NRG’s Role in the Initial Establishment of to the transformation of radial glial cells into astrocytes. -

Pre-Oligodendrocytes from Adult Human CNS
The Journal of Neuroscience, April 1992, 12(4): 1538-l 547 Pre-Oligodendrocytes from Adult Human CNS Regina C. Armstrong,lJ Henry H. Dorn, l,b Conrad V. Kufta,* Emily Friedman,3 and Monique E. Dubois-Dalcq’ ‘Laboratory of Viral and Molecular Pathogenesis, and %urgical Neurology Branch, National Institute of Neurological Disorders and Stroke, Bethesda, Maryland 20892 and 3Department of Neurosurgery, University of Pennsylvania, Philadelphia, Pennsylvania 19104-3246 CNS remyelination and functional recovery often occur after Rapid and efficient neurotransmission is dependent upon the experimental demyelination in adult rodents. This has been electrical insulating capacity of the myelin sheath around axons attributed to the ability of mature oligodendrocytes and/or (reviewed in Ritchie, 1984a,b). Nerve conduction is impaired their precursor cells to divide and regenerate in response after loss of the myelin sheath and results in severe neurological to signals in demyelinating lesions. To determine whether dysfunction in human demyelinating diseases such as multiple oligodendrocyte precursor cells exist in the adult human sclerosis (MS). Remyelination can occur in the CNS of MS CNS, we have cultured white matter from patients under- patients but appears to be limited (Perier and Gregoire, 1965; going partial temporal lobe resection for intractable epilep- Prineas et al., 1984). Studies of acute MS cases have revealed sy. These cultures contained a population of process-bear- that recent demyelinating lesions can exhibit remyelination that ing cells that expressed antigens recognized by the 04 appears to correlate with the generation of new oligodendrocytes monoclonal antibody, but these cells did not express galac- (Prineas et al., 1984; Raine et al., 1988). -

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response
Supplementary Online Content Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. Published online May 28, 2015. doi:10.1001/jamaoncol.2015.1313 eMethods eFigure 1. A schematic of the IPM Computational Pipeline eFigure 2. Tumor purity analysis eFigure 3. Tumor purity estimates from Pathology team versus computationally (CLONET) estimated tumor purities values for frozen tumor specimens (Spearman correlation 0.2765327, p- value = 0.03561) eFigure 4. Sequencing metrics Fresh/frozen vs. FFPE tissue eFigure 5. Somatic copy number alteration profiles by tumor type at cytogenetic map location resolution; for each cytogenetic map location the mean genes aberration frequency is reported eFigure 6. The 20 most frequently aberrant genes with respect to copy number gains/losses detected per tumor type eFigure 7. Top 50 genes with focal and large scale copy number gains (A) and losses (B) across the cohort eFigure 8. Summary of total number of copy number alterations across PM tumors eFigure 9. An example of tumor evolution looking at serial biopsies from PM222, a patient with metastatic bladder carcinoma eFigure 10. PM12 somatic mutations by coverage and allele frequency (A) and (B) mutation correlation between primary (y- axis) and brain metastasis (x-axis) eFigure 11. Point mutations across 5 metastatic sites of a 55 year old patient with metastatic prostate cancer at time of rapid autopsy eFigure 12. CT scans from patient PM137, a patient with recurrent platinum refractory metastatic urothelial carcinoma eFigure 13. Tracking tumor genomics between primary and metastatic samples from patient PM12 eFigure 14. -

View Software
TLR4-activated microglia have divergent effects on oligodendrocyte lineage cells DISSERTATION Presented in partial fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University Evan Zachary Goldstein, BA Neuroscience Graduate Program The Ohio State University 2016 Dissertation Committee: Dr. Dana McTigue, Advisor Dr. Phillip Popovich Dr. Jonathan Godbout Dr. Courtney DeVries Copyright by Evan Zachary Goldstein 2016 i Abstract Myelin accelerates action potential conduction velocity and provides essential metabolic support for axons. Unfortunately, myelin and myelinating cells are often vulnerable to injury or disease, resulting in myelin damage, which in turn can lead to axon dysfunction, overt pathology and neurological impairment. Inflammation is a common component of CNS trauma and disease, and therefore an active inflammatory response is often considered deleterious to myelin health. While inflammation can certainly damage myelin, inflammatory processes also benefit oligodendrocyte (OL) lineage progression and myelin repair. Consistent with the divergent nature of inflammation, intraspinal toll-like receptor 4 (TLR4) activation, an innate immune pathway, kills OL lineage cells, but also initiates oligodendrogenesis. Soluble factors produced by TLR4-activated microglia can reproduce these effects in vitro, however the exact factors are unknown. To determine what microglial factors might contribute to TLR4-induced OL loss and oligodendrogenesis, mRNA of factors known to affect OL lineage cells was quantified in TLR4-activated microglia and spinal cords (chapter 2). Results indicate that TLR4-activated microglia transcribe numerous factors that induce OL loss, OL progenitor cell (OPC) proliferation and OPC differentiation. However, some factors upregulated after intraspinal TLR4 activation were not ii upregulated by microglia, suggesting that other cell types contribute to transcriptional changes in vivo. -

Wo2017/132291
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date W O 2017/132291 A l 3 August 2017 (03.08.2017) P O P C T (51) International Patent Classification: [US/US]; 77 Massachusetts Avenue, Cambridge, MA A61K 48/00 (2006.01) C12Q 1/68 (2006.01) 02139 (US). THE GENERAL HOSPITAL CORPORA¬ A61K 39/395 (2006.01) G01N 33/574 (2006.01) TION [US/US]; 55 Fruit Street, Boston, MA 021 14 (US). C12N 15/11 (2006.01) (72) Inventors; and (21) International Application Number: (71) Applicants : REGEV, Aviv [US/US]; 415 Main Street, PCT/US2017/014995 Cambridge, MA 02142 (US). BERNSTEIN, Bradley [US/US]; 55 Fruit Street, Boston, MA 021 14 (US). (22) International Filing Date: TIROSH, Itay [US/US]; 415 Main Street, Cambridge, 25 January 20 17 (25.01 .2017) MA 02142 (US). SUVA, Mario [US/US]; 55 Fruit Street, (25) Filing Language: English Bostn, MA 02144 (US). ROZENBALTT-ROSEN, Orit [US/US]; 415 Main Street, Cambridge, MA 02142 (US). (26) Publication Language: English (74) Agent: NIX, F., Brent; Johnson, Marcou & Isaacs, LLC, (30) Priority Data: 317A East Liberty St., Savannah, GA 31401 (US). 62/286,850 25 January 2016 (25.01.2016) US 62/437,558 2 1 December 201 6 (21. 12.2016) US (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (71) Applicants: THE BROAD INSTITUTE, INC. [US/US]; AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 415 Main Street, Cambridge, MA 02142 (US). -
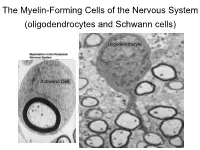
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Open Full Page
Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 RESEARCH ARTICLE Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer Hannah Carter 1 , 2 , 3 , 4 , Rachel Marty 5 , Matan Hofree 6 , Andrew M. Gross 5 , James Jensen 5 , Kathleen M. Fisch1,2,3,7 , Xingyu Wu 2 , Christopher DeBoever 5 , Eric L. Van Nostrand 4,8 , Yan Song 4,8 , Emily Wheeler 4,8 , Jason F. Kreisberg 1,3 , Scott M. Lippman 2 , Gene W. Yeo 4,8 , J. Silvio Gutkind 2 , 3 , and Trey Ideker 1 , 2 , 3 , 4 , 5,6 Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2017 American Association for Cancer Research. Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 ABSTRACT Recent studies have characterized the extensive somatic alterations that arise dur- ing cancer. However, the somatic evolution of a tumor may be signifi cantly affected by inherited polymorphisms carried in the germline. Here, we analyze genomic data for 5,954 tumors to reveal and systematically validate 412 genetic interactions between germline polymorphisms and major somatic events, including tumor formation in specifi c tissues and alteration of specifi c cancer genes. Among germline–somatic interactions, we found germline variants in RBFOX1 that increased incidence of SF3B1 somatic mutation by 8-fold via functional alterations in RNA splicing. Similarly, 19p13.3 variants were associated with a 4-fold increased likelihood of somatic mutations in PTEN. In support of this associ- ation, we found that PTEN knockdown sensitizes the MTOR pathway to high expression of the 19p13.3 gene GNA11 . -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function.