Deficient Mutant Identifies Novel Alterations in Gene Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download (PDF)
ANALYTICAL SCIENCES NOVEMBER 2020, VOL. 36 1 2020 © The Japan Society for Analytical Chemistry Supporting Information Fig. S1 Detailed MS/MS data of myoglobin. 17 2 ANALYTICAL SCIENCES NOVEMBER 2020, VOL. 36 Table S1 : The protein names (antigens) identified by pH 2.0 solution in the eluted-fraction. These proteins were identified one or more out of six analyses. Accession Description P08908 5-hydroxytryptamine receptor 1A OS=Homo sapiens GN=HTR1A PE=1 SV=3 - [5HT1A_HUMAN] Q9NRR6 72 kDa inositol polyphosphate 5-phosphatase OS=Homo sapiens GN=INPP5E PE=1 SV=2 - [INP5E_HUMAN] P82987 ADAMTS-like protein 3 OS=Homo sapiens GN=ADAMTSL3 PE=2 SV=4 - [ATL3_HUMAN] Q9Y6K8 Adenylate kinase isoenzyme 5 OS=Homo sapiens GN=AK5 PE=1 SV=2 - [KAD5_HUMAN] P02763 Alpha-1-acid glycoprotein 1 OS=Homo sapiens GN=ORM1 PE=1 SV=1 - [A1AG1_HUMAN] P19652 Alpha-1-acid glycoprotein 2 OS=Homo sapiens GN=ORM2 PE=1 SV=2 - [A1AG2_HUMAN] P01011 Alpha-1-antichymotrypsin OS=Homo sapiens GN=SERPINA3 PE=1 SV=2 - [AACT_HUMAN] P01009 Alpha-1-antitrypsin OS=Homo sapiens GN=SERPINA1 PE=1 SV=3 - [A1AT_HUMAN] P04217 Alpha-1B-glycoprotein OS=Homo sapiens GN=A1BG PE=1 SV=4 - [A1BG_HUMAN] P08697 Alpha-2-antiplasmin OS=Homo sapiens GN=SERPINF2 PE=1 SV=3 - [A2AP_HUMAN] P02765 Alpha-2-HS-glycoprotein OS=Homo sapiens GN=AHSG PE=1 SV=1 - [FETUA_HUMAN] P01023 Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 - [A2MG_HUMAN] P01019 Angiotensinogen OS=Homo sapiens GN=AGT PE=1 SV=1 - [ANGT_HUMAN] Q9NQ90 Anoctamin-2 OS=Homo sapiens GN=ANO2 PE=1 SV=2 - [ANO2_HUMAN] P01008 Antithrombin-III -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Diversification of the Caenorhabditis Heat Shock Response by Helitron Transposable Elements Jacob M Garrigues, Brian V Tsu, Matthew D Daugherty, Amy E Pasquinelli*
RESEARCH ARTICLE Diversification of the Caenorhabditis heat shock response by Helitron transposable elements Jacob M Garrigues, Brian V Tsu, Matthew D Daugherty, Amy E Pasquinelli* Division of Biology, University of California, San Diego, San Diego, United States Abstract Heat Shock Factor 1 (HSF-1) is a key regulator of the heat shock response (HSR). Upon heat shock, HSF-1 binds well-conserved motifs, called Heat Shock Elements (HSEs), and drives expression of genes important for cellular protection during this stress. Remarkably, we found that substantial numbers of HSEs in multiple Caenorhabditis species reside within Helitrons, a type of DNA transposon. Consistent with Helitron-embedded HSEs being functional, upon heat shock they display increased HSF-1 and RNA polymerase II occupancy and up-regulation of nearby genes in C. elegans. Interestingly, we found that different genes appear to be incorporated into the HSR by species-specific Helitron insertions in C. elegans and C. briggsae and by strain-specific insertions among different wild isolates of C. elegans. Our studies uncover previously unidentified targets of HSF-1 and show that Helitron insertions are responsible for rewiring and diversifying the Caenorhabditis HSR. Introduction Heat Shock Factor 1 (HSF-1) is a highly conserved transcription factor that serves as a key regulator of the heat shock response (HSR) (Vihervaara et al., 2018). In response to elevated temperatures, HSF-1 binds well-conserved motifs, termed heat shock elements (HSEs), and drives the transcription *For correspondence: of genes important for mitigating the proteotoxic effects of heat stress. For example, HSF-1 pro- [email protected] motes the expression of heat-shock proteins (HSPs) that act as chaperones to prevent HS-induced misfolding and aggregation of proteins (Vihervaara et al., 2018). -

Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response
Supplementary Online Content Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. Published online May 28, 2015. doi:10.1001/jamaoncol.2015.1313 eMethods eFigure 1. A schematic of the IPM Computational Pipeline eFigure 2. Tumor purity analysis eFigure 3. Tumor purity estimates from Pathology team versus computationally (CLONET) estimated tumor purities values for frozen tumor specimens (Spearman correlation 0.2765327, p- value = 0.03561) eFigure 4. Sequencing metrics Fresh/frozen vs. FFPE tissue eFigure 5. Somatic copy number alteration profiles by tumor type at cytogenetic map location resolution; for each cytogenetic map location the mean genes aberration frequency is reported eFigure 6. The 20 most frequently aberrant genes with respect to copy number gains/losses detected per tumor type eFigure 7. Top 50 genes with focal and large scale copy number gains (A) and losses (B) across the cohort eFigure 8. Summary of total number of copy number alterations across PM tumors eFigure 9. An example of tumor evolution looking at serial biopsies from PM222, a patient with metastatic bladder carcinoma eFigure 10. PM12 somatic mutations by coverage and allele frequency (A) and (B) mutation correlation between primary (y- axis) and brain metastasis (x-axis) eFigure 11. Point mutations across 5 metastatic sites of a 55 year old patient with metastatic prostate cancer at time of rapid autopsy eFigure 12. CT scans from patient PM137, a patient with recurrent platinum refractory metastatic urothelial carcinoma eFigure 13. Tracking tumor genomics between primary and metastatic samples from patient PM12 eFigure 14. -

Wo2017/132291
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date W O 2017/132291 A l 3 August 2017 (03.08.2017) P O P C T (51) International Patent Classification: [US/US]; 77 Massachusetts Avenue, Cambridge, MA A61K 48/00 (2006.01) C12Q 1/68 (2006.01) 02139 (US). THE GENERAL HOSPITAL CORPORA¬ A61K 39/395 (2006.01) G01N 33/574 (2006.01) TION [US/US]; 55 Fruit Street, Boston, MA 021 14 (US). C12N 15/11 (2006.01) (72) Inventors; and (21) International Application Number: (71) Applicants : REGEV, Aviv [US/US]; 415 Main Street, PCT/US2017/014995 Cambridge, MA 02142 (US). BERNSTEIN, Bradley [US/US]; 55 Fruit Street, Boston, MA 021 14 (US). (22) International Filing Date: TIROSH, Itay [US/US]; 415 Main Street, Cambridge, 25 January 20 17 (25.01 .2017) MA 02142 (US). SUVA, Mario [US/US]; 55 Fruit Street, (25) Filing Language: English Bostn, MA 02144 (US). ROZENBALTT-ROSEN, Orit [US/US]; 415 Main Street, Cambridge, MA 02142 (US). (26) Publication Language: English (74) Agent: NIX, F., Brent; Johnson, Marcou & Isaacs, LLC, (30) Priority Data: 317A East Liberty St., Savannah, GA 31401 (US). 62/286,850 25 January 2016 (25.01.2016) US 62/437,558 2 1 December 201 6 (21. 12.2016) US (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (71) Applicants: THE BROAD INSTITUTE, INC. [US/US]; AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 415 Main Street, Cambridge, MA 02142 (US). -

Open Full Page
Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 RESEARCH ARTICLE Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer Hannah Carter 1 , 2 , 3 , 4 , Rachel Marty 5 , Matan Hofree 6 , Andrew M. Gross 5 , James Jensen 5 , Kathleen M. Fisch1,2,3,7 , Xingyu Wu 2 , Christopher DeBoever 5 , Eric L. Van Nostrand 4,8 , Yan Song 4,8 , Emily Wheeler 4,8 , Jason F. Kreisberg 1,3 , Scott M. Lippman 2 , Gene W. Yeo 4,8 , J. Silvio Gutkind 2 , 3 , and Trey Ideker 1 , 2 , 3 , 4 , 5,6 Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2017 American Association for Cancer Research. Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 ABSTRACT Recent studies have characterized the extensive somatic alterations that arise dur- ing cancer. However, the somatic evolution of a tumor may be signifi cantly affected by inherited polymorphisms carried in the germline. Here, we analyze genomic data for 5,954 tumors to reveal and systematically validate 412 genetic interactions between germline polymorphisms and major somatic events, including tumor formation in specifi c tissues and alteration of specifi c cancer genes. Among germline–somatic interactions, we found germline variants in RBFOX1 that increased incidence of SF3B1 somatic mutation by 8-fold via functional alterations in RNA splicing. Similarly, 19p13.3 variants were associated with a 4-fold increased likelihood of somatic mutations in PTEN. In support of this associ- ation, we found that PTEN knockdown sensitizes the MTOR pathway to high expression of the 19p13.3 gene GNA11 . -

IDENTIFYING a ROLE for HEAT SHOCK PROTEINS in SCHISTOSOMA MANSONI by KENJI ISHIDA CASE WESTERN RESERVE UNIVERSITY
IDENTIFYING A ROLE FOR HEAT SHOCK PROTEINS IN SCHISTOSOMA MANSONI by KENJI ISHIDA Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Biology CASE WESTERN RESERVE UNIVERSITY August 2017 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Kenji Ishida candidate for the degree of Doctor of Philosophy Committee Chair Michael Benard Committee Member Ronald Blanton Committee Member Christopher Cullis Committee Member Claudia Mieko Mizutani Committee Member Emmitt R. Jolly Date of Defense April 26, 2017 *We also certify that written approval has been obtained for any proprietary material contained therin. Table of Contents Table of Contents Table of Contents ............................................................................................................. iii List of Figures .................................................................................................................. vii List of Abbreviations ...................................................................................................... viii Abstract ........................................................................................................................... xiv Chapter 1: Introduction ................................................................................................... 1 1.1 Purpose ...................................................................................................................... 1 1.2 Schistosomiasis -

ANG II Promotes IGF-IIR Expression and Cardiomyocyte Apoptosis by Inhibiting HSF1 Via JNK Activation and SIRT1 Degradation
Cell Death and Differentiation (2014) 21, 1262–1274 & 2014 Macmillan Publishers Limited All rights reserved 1350-9047/14 www.nature.com/cdd ANG II promotes IGF-IIR expression and cardiomyocyte apoptosis by inhibiting HSF1 via JNK activation and SIRT1 degradation C-Y Huang1, W-W Kuo2, Y-L Yeh3,4, T-J Ho5,6, J-Y Lin7,8, D-Y Lin1, C-H Chu1, F-J Tsai6, C-H Tsai9 and C-Y Huang*,1,6,10 Hypertension-induced cardiac hypertrophy and apoptosis are major characteristics of early-stage heart failure. Our previous studies found that the activation of insulin-like growth factor receptor II (IGF-IIR) signaling was critical for hypertensive angiotensin II (ANG II)-induced cardiomyocyte apoptosis. However, the detailed mechanism by which ANG II regulates IGF-IIR in heart cells remains elusive. In this study, we found that ANG II activated its downstream kinase JNK to increase IGF-IIR expression through the ANG II receptor angiotensin type 1 receptor. JNK activation subsequently led to sirtuin 1 (SIRT1) degradation via the proteasome, thus preventing SIRT1 from deacetylating heat-shock transcription factor 1 (HSF1). The resulting increase in the acetylation of HSF1 impaired its ability to bind to the IGF-IIR promoter region (nt À 748 to À 585). HSF1 protected cardiomyocytes by acting as a repressor of IGF-IIR gene expression, and ANG II diminished this HSF1-mediated repression through enhanced acetylation, thus activating the IGF-IIR apoptosis pathway. Taken together, these results suggest that HSF1 represses IGF-IIR gene expression to protect cardiomyocytes. ANG II activates JNK to degrade SIRT1, resulting in HSF1 acetylation, which induces IGF-IIR expression and eventually results in cardiac hypertrophy and apoptosis. -

Table SII. Significantly Differentially Expressed Mrnas of GSE23558 Data Series with the Criteria of Adjusted P<0.05 And
Table SII. Significantly differentially expressed mRNAs of GSE23558 data series with the criteria of adjusted P<0.05 and logFC>1.5. Probe ID Adjusted P-value logFC Gene symbol Gene title A_23_P157793 1.52x10-5 6.91 CA9 carbonic anhydrase 9 A_23_P161698 1.14x10-4 5.86 MMP3 matrix metallopeptidase 3 A_23_P25150 1.49x10-9 5.67 HOXC9 homeobox C9 A_23_P13094 3.26x10-4 5.56 MMP10 matrix metallopeptidase 10 A_23_P48570 2.36x10-5 5.48 DHRS2 dehydrogenase A_23_P125278 3.03x10-3 5.40 CXCL11 C-X-C motif chemokine ligand 11 A_23_P321501 1.63x10-5 5.38 DHRS2 dehydrogenase A_23_P431388 2.27x10-6 5.33 SPOCD1 SPOC domain containing 1 A_24_P20607 5.13x10-4 5.32 CXCL11 C-X-C motif chemokine ligand 11 A_24_P11061 3.70x10-3 5.30 CSAG1 chondrosarcoma associated gene 1 A_23_P87700 1.03x10-4 5.25 MFAP5 microfibrillar associated protein 5 A_23_P150979 1.81x10-2 5.25 MUCL1 mucin like 1 A_23_P1691 2.71x10-8 5.12 MMP1 matrix metallopeptidase 1 A_23_P350005 2.53x10-4 5.12 TRIML2 tripartite motif family like 2 A_24_P303091 1.23x10-3 4.99 CXCL10 C-X-C motif chemokine ligand 10 A_24_P923612 1.60x10-5 4.95 PTHLH parathyroid hormone like hormone A_23_P7313 6.03x10-5 4.94 SPP1 secreted phosphoprotein 1 A_23_P122924 2.45x10-8 4.93 INHBA inhibin A subunit A_32_P155460 6.56x10-3 4.91 PICSAR P38 inhibited cutaneous squamous cell carcinoma associated lincRNA A_24_P686965 8.75x10-7 4.82 SH2D5 SH2 domain containing 5 A_23_P105475 7.74x10-3 4.70 SLCO1B3 solute carrier organic anion transporter family member 1B3 A_24_P85099 4.82x10-5 4.67 HMGA2 high mobility group AT-hook 2 A_24_P101651 -
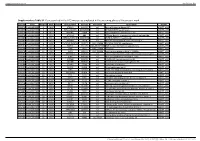
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Novel IQCE Variations Confirm Its Role in Postaxial Polydactyly and Cause
Novel IQCE variations confirm its role in postaxial polydactyly and cause ciliary defect phenotype in zebrafish Alejandro Estrada-Cuzcano, Christelle Etard, Clarisse Delvallée, Corinne Stoetzel, Elise Schaefer, Sophie Scheidecker, Véronique Geoffroy, Aline Schneider, Fouzia Studer, Francesca Mattioli, et al. To cite this version: Alejandro Estrada-Cuzcano, Christelle Etard, Clarisse Delvallée, Corinne Stoetzel, Elise Schaefer, et al.. Novel IQCE variations confirm its role in postaxial polydactyly and cause ciliary defect phenotype in zebrafish. Human Mutation, Wiley, 2019, 10.1002/humu.23924. hal-02304111 HAL Id: hal-02304111 https://hal.archives-ouvertes.fr/hal-02304111 Submitted on 2 Oct 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Page 1 of 125 Human Mutation Novel IQCE variations confirm its role in postaxial polydactyly and cause ciliary defect phenotype in zebrafish. Alejandro Estrada-Cuzcano1*, Christelle Etard2*, Clarisse Delvallée1*, Corinne Stoetzel1, Elise Schaefer1,3, Sophie Scheidecker1,4, Véronique Geoffroy1, Aline Schneider1, Fouzia Studer5, Francesca Mattioli6, Kirsley Chennen1,7, Sabine Sigaudy8, Damien Plassard9, Olivier Poch7, Amélie Piton4,6, Uwe Strahle2, Jean Muller1,4* and Hélène Dollfus1,3,5* 1. Laboratoire de Génétique médicale, UMR_S INSERM U1112, IGMA, Faculté de Médecine FMTS, Université de Strasbourg, Strasbourg, France. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature.