Blueprint Genetics Non-Syndromic Hearing Loss Panel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Age-Related Hearing Loss
U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES ∙ National Institutes of Health NIDCD Fact Sheet | Hearing and Balance Age-Related Hearing Loss What is age-related hearing loss? The auditory system Age-related hearing loss (presbycusis) is the loss of hearing that gradually occurs in most of us as we grow older. It is one of the most common conditions affecting older and elderly adults. Approximately one in three people in the United States between the ages of 65 and 74 has hearing loss, and nearly half of those older than 75 have difficulty hearing. Having trouble hearing can make it hard to understand and follow a doctor’s advice, respond to warnings, and hear phones, doorbells, and smoke alarms. Hearing loss can also make it hard to enjoy talking with family and friends, leading to feelings of isolation. Age-related hearing loss most often occurs in both ears, affecting them equally. Because the loss is gradual, if you have age-related hearing loss you Credit: NIH Medical Arts may not realize that you’ve lost some of your ability to hear. How do we hear? There are many causes of age-related hearing Hearing depends on a series of events that change loss. Most commonly, it arises from changes in sound waves in the air into electrical signals. Your the inner ear as we age, but it can also result auditory nerve then carries these signals to your from changes in the middle ear, or from complex brain through a complex series of steps. changes along the nerve pathways from the ear 1. -
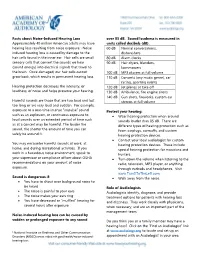
Facts About Noise-Induced Hearing Loss Over 85 Db
Facts about Noise-Induced Hearing Loss over 85 dB. Sound loudness is measured in Approximately 40 million American adults may have units called decibels (dB). hearing loss resulting from noise exposure.1 Noise- 60 dB Normal conversations, induced hearing loss is caused by damage to the dishwashers hair cells found in the inner ear. Hair cells are small 80 dB Alarm clocks sensory cells that convert the sounds we hear 90 dB Hair dryers, blenders, (sound energy) into electrical signals that travel to lawnmowers the brain. Once damaged, our hair cells cannot 100 dB MP3 players at full volume grow back, which results in permanent hearing loss. 110 dB Concerts (any music genre), car racing, sporting events Hearing protection decreases the intensity, or 120 dB Jet planes at take off loudness, of noise and helps preserve your hearing. 130 dB Ambulance, fire engine sirens 140 dB Gun shots, fireworks, custom car Harmful sounds are those that are too loud and last stereos at full volume too long or are very loud and sudden. For example, exposure to a one-time intense “impulse” sound Protect your hearing: such as an explosion, or continuous exposure to • Wear hearing protection when around loud sounds over an extended period of time such sounds louder than 85 dB. There are as at a concert may be harmful. The louder the different types of hearing protection such as sound, the shorter the amount of time you can foam earplugs, earmuffs, and custom safely be around it. hearing protection devices • Contact your local audiologist for custom You may encounter harmful sounds at work, at hearing protection devices. -

Hearing Loss
Randal W. Swenson, M.D. Joshua G. Yorgason, M.D. David K. Palmer, M.D. Wesley R. Brown, M.D. John E. Butler, M.D. Nancy J. Stevenson, PA-C Justin D. Gull, M.D. ENT SPECIALISTS Kristin G. Hoopes, PA-C www.entslc.com Hearing Loss Approximately one in ten persons in the United may result from blockage of the ear canal (wax), States has some degree of hearing loss. Hearing is from a perforation (hole) in the ear drum, or from measured in decibels (dB), and a hearing level of 0- infection or disease of any of the three middle ear 25 dB is considered normal hearing. Your level is: bones. With a conductive loss only, the patient will never go deaf, but will always be able to hear, either Right ear _______ dB Left ear _______dB with reconstructive ear surgery or by use of a properly fitted hearing aid. Some patients who are Hearing Severity / % Loss not candidates for surgery, may benefit from a new 25 dB (normal).….0% 65dB(Severe)……...60% technology, the Baha (bone-anchored hearing aid). 35 dB (mild)……..15% 75dB(Severe)……...75% When there is a problem with the inner ear or 45 dB (moderate)..30% >85dB (Profound)..>90% nerve of hearing, a sensori-neural hearing loss occurs. This is most commonly from normal aging, Normal speech discrimination is 88-100%. Yours is: is usually worse in high frequencies, and can progress to total deafness. Noise exposure is another Right ear _______ % Left ear_______% common cause of high frequency hearing loss. Patients with sensori-neural hearing loss usually complain of difficulty hearing in loud environments. -

Preventing Hearing Loss Caused by Chemical (Ototoxicity) and Noise Exposure
Preventing Hearing Loss Caused by Chemical (Ototoxicity) and Noise Exposure Safety and Health Information Bulletin SHIB 03-08-2018 DHHS (NIOSH) Publication No. 2018-124 Introduction Millions of workers are exposed to noise in the workplace every day and when uncontrolled, noise exposure may cause permanent hearing loss. Research demonstrates exposure to certain chemicals, called ototoxicants, may cause hearing loss or balance problems, regardless of noise exposure. Substances including certain pesticides, solvents, and pharmaceuticals that contain ototoxicants can negatively affect how the ear functions, causing hearing loss, and/or affect balance. Source/Copyright: OSHA The risk of hearing loss is increased when workers are exposed to these chemicals while working around elevated noise levels. This combination often results in hearing loss that can be temporary or permanent, depending on the level of noise, the dose of the chemical, and the duration of the exposure. This hearing impairment affects many occupations and industries, from machinists to firefighters. Effects on Hearing Harmful exposure to ototoxicants may occur through inhalation, ingestion, or skin absorption. Health effects caused by ototoxic chemicals vary based on exposure frequency, intensity, duration, workplace exposure to other hazards, and individual factors such as age. Effects may be temporary or permanent, can affect hearing sensitivity and result in a standard threshold shift. Since chemicals can affect central portions of the auditory system (e.g., nerves or nuclei in the central nervous system, the pathways to the brain or in the brain itself), not only do sounds need to be louder to be detected, but also they lose clarity. Specifically, speech discrimination dysfunction, the ability to hear voices separately from background noise, may occur and involve: . -

Differential Diagnosis and Treatment of Hearing Loss JON E
Differential Diagnosis and Treatment of Hearing Loss JON E. ISAACSON, M.D., and NEIL M. VORA, M.D., Milton S. Hershey Medical Center, Hershey, Pennsylvania Hearing loss is a common problem that can occur at any age and makes verbal communication difficult. The ear is divided anatomically into three sections (external, middle, and inner), and pathology contributing to hearing loss may strike one or more sections. Hearing loss can be cat- egorized as conductive, sensorineural, or both. Leading causes of conductive hearing loss include cerumen impaction, otitis media, and otosclerosis. Leading causes of sensorineural hear- ing loss include inherited disorders, noise exposure, and presbycusis. An understanding of the indications for medical management, surgical treatment, and amplification can help the family physician provide more effective care for these patients. (Am Fam Physician 2003;68:1125-32. Copyright© 2003 American Academy of Family Physicians) ore than 28 million Amer- tive, the sound will be heard best in the icans have some degree of affected ear. If the loss is sensorineural, the hearing impairment. The sound will be heard best in the normal ear. differential diagnosis of The sound remains midline in patients with hearing loss can be sim- normal hearing. Mplified by considering the three major cate- The Rinne test compares air conduction gories of loss. Conductive hearing loss occurs with bone conduction. The tuning fork is when sound conduction is impeded through struck softly and placed on the mastoid bone the external ear, the middle ear, or both. Sen- (bone conduction). When the patient no sorineural hearing loss occurs when there is a longer can hear the sound, the tuning fork is problem within the cochlea or the neural placed adjacent to the ear canal (air conduc- pathway to the auditory cortex. -

UNDERSTANDING an AUDIOGRAM Marni L
SERVING STUDENTS WHO ARE HARD OF HEARING UNDERSTANDING AN AUDIOGRAM Marni L. Johnson University of South Dakota SUMMARY from 125 to 8000 Hz. The normal hearing listener can typically hear sounds as soft as 0 dB HL if one is present, can be determined by reading an and when sounds are above 100 dB HL they are audiogram. The type of hearing loss is determined by generally considered to be uncomfortably loud. comparing auditory thresholds obtained using head- phones or insert earphones (air-conduction thresholds) KEY CONCEPTS to those obtained using a bone oscillator (bone-conduc- tion thresholds). By itself, the audiogram cannot tell us Conductive hearing losses (CHL) how an individual will perform in the real world. While CHL are characterized by a reduction in hearing ability tests of speech perception in quiet and in noise can despite a normal functioning cochlea (inner ear). This greatly enhance the diagnostic value of the audiogram, type of hearing loss is caused by impaired sound the results obtained in a sound booth do not always transmission through the ear canal, eardrum, and/ translate directly to how an individual will perform in or ossicular chain. Conductive hearing losses are the real world infections and wax impaction are two common causes KEY TERMINOLOGY of this type of hearing loss. In conductive hearing losses, air conduction thresholds are abnormal, bone Audiogram conduction thresholds are normal, and an air-bone gap is present. representing the softest sounds that a person can Sensorineural hearing losses (SNHL) Decibel (dB) Decibel refers to the loudness of sounds. A sound SNHL are characterized by a reduction in hearing low in dB is perceived as soft and a sound high in dB ability due to disorders involving the cochlea and/or is perceived as loud. -

8 Hearing Measurement
8 HEARING MEASUREMENT John R. Franks, Ph.D. Chief, Hearing Loss Prevention Section Engineering and Physical Hazards Branch Division of Applied Research and Technology National Institute for Occupational Safety and Health Robert A. Taft Laboratories 4676 Columbia Parkway Cincinnati, Ohio 45226-1998 USA [email protected] 8.1. INTRODUCTION (RATIONALE FOR AUDIOMETRY) The audiogram is a picture of how a person hears at a given place and time under given conditions. The audiogram may be used to describe the hearing of a person for the various frequencies tested. It may be used to calculate the amount of hearing handicap a person has. And, it may be used as a tool to determine the cause of a person’s hearing loss. Audiograms may be obtained in many ways; e.g., by using pure tones via air conduction or bone conduction for behavioral testing or by using tone pips to generate auditory brainstem responses. The audiogram is a most unusual biometric test. It is often incorrectly compared to a vision test. In the audiogram, the goal is to determine the lowest signal level a person can hear. In the case of a vision test, the person reads the smallest size of print that he or she can see, the auditory equivalent of identifying the least perceptible difference between two sounds. In most occupational and medical settings, this requires the listener to respond to very low levels of sounds that he or she does not hear in normal day-to-day life. A vision test analogous to an audiogram would require a person to sit in a totally darkened room and be tested for the lowest luminosity light of various colors, red to blue, that can be seen. -

Conductive Hearing Loss
tm Conductive Hearing Loss 1. What is auditory conductive hearing loss? Conductive hearing loss is the most common cause of hearing impair- ment- especially in children. The “conductive” component of the problem describes the blockage of sounds from reaching the sensory cells of the inner ear. In conductive hearing loss, the inner ear functions normally, but sound vibrations are blocked from passage through the ear canal, ear drum or across the tiny bones located in the middle ear. Patients with conduc- tive hearing loss hear bone-conducted sounds presented with a small vibra- tor to the skull better (louder) than sounds presented through earphones. Conductive hearing loss is usually mild to moderate in degree, may occur in one or both ears at the same time, and in most cases is correctable by relatively minor medical or surgical treatments. More significant conduc- tive hearing loss may be associated with skull and/or facial malformations which require technical surgery to correct. 2. What issues are at the forefront of conductive hearing loss? Diagnosis of conductive hearing loss is not difficult in the experienced hands of an audiologist or otolaryngologist (Ear-Nose-Throat Physician). Special hearing test techniques are available to quantify the degree of the conductive hearing loss such as comparing bone-conduction thresholds with air-conduction thresholds, tympanometry and acoustic reflex mea- surements, and otoacoustic emissions testing. Your audiologist should use all these techniques to verify and delineate the conductive hearing. These tests are more difficult to administer to very young children and require Communication Considerations A-ZTM Conductive Hearing Loss an experienced audiologist experienced with pediatric patients. -

Analysis of Chronic Tinnitus in Noise-Induced Hearing Loss and Presbycusis
Journal of Clinical Medicine Article Analysis of Chronic Tinnitus in Noise-Induced Hearing Loss and Presbycusis Hee Jin Kang 1, Dae Woong Kang 1, Sung Su Kim 2, Tong In Oh 3 , Sang Hoon Kim 1 and Seung Geun Yeo 1,* 1 Department of Otolaryngology-Head & Neck Surgery, School of Medicine, Kyung Hee University, Seoul 02447, Korea; [email protected] (H.J.K.); [email protected] (D.W.K.); [email protected] (S.H.K.) 2 Medical Research Center for Bioreaction to Reactive Oxygen Species and Biomedical Science Institute, School of Medicine, Graduate School, Kyung Hee University, Seoul 02447, Korea; [email protected] 3 Department of Biomedical Engineering, College of Medicine, Kyung Hee University, Seoul 02447, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-2-958-8980; Fax: +82-2-958-8470 Abstract: Introduction: The most frequent causes of tinnitus associated with hearing loss are noise- induced hearing loss and presbycusis. The mechanism of tinnitus is not yet clear, although several hypotheses have been suggested. Therefore, we aimed to analyze characteristics of chronic tinnitus between noise-induced hearing loss and presbycusis. Materials and Methods: This paper is a retrospective chart review and outpatient clinic-based study of 248 patients with chronic tinnitus from 2015 to 2020 with noise-induced or presbycusis. Pure tone audiometry (PTA), auditory brainstem response (ABR), distortion product otoacoustic emissions (DPOAE), transient evoked otoacoustic emissions (TEOAE), and tinnitograms were conducted. Results: PTA showed that hearing thresholds at all frequencies were higher in patients with noise-induced hearing loss than the presbycusis group. Citation: Kang, H.J.; Kang, D.W.; ABR tests showed that patients with presbycusis had longer wave I and III latencies (p < 0.05 each) Kim, S.S.; Oh, T.I.; Kim, S.H.; Yeo, S.G. -

Noise-Induced Hearing Loss (NIHL)
Noise-Induced Hearing Loss (NIHL) About 12.5 percent of children and teens have lasting hearing loss from contact with very loud noise (Sight and Hearing Association, 2013). Why does loud noise cause hearing loss? There are 15,000 to 20,000 tiny cells (cilia) in the inner ear that send sounds to the brain. Hearing loss can result when these cilia are damaged. To picture what happens when these cells are exposed to too much noise, think about when you walk on fresh grass. When you walk on it lightly only a few times it bounces back to its original shape. If you walk on grass often, or you crush it, it becomes matted down and will not bounce back. Exposure to sounds that are loud enough or occur long enough can damage the ears’ cilia so that they can no longer bounce back into shape and can no longer send sound to the brain. This is the cause of noise-induced hearing loss (NIHL). Frequency is the tone of a sound. The frequency of a sound can range from very low to very high tones. At first, NIHL can make it hard to hear high tones. This can cause problems hearing speech sounds like “s”. Background noise often makes hearing these sounds even harder to hear. NIHL slowly progresses into lower tones. Once these are affected, you may have problems hearing people when they speak. What kind of noise is too loud? The noise is too loud if you have to raise your voice to talk to a person who is only an arm’s length away. -

Conductive Hearing Loss
Conductive hearing loss Information for patients This sheet answers common questions about conductive hearing loss. If you would like further information, or have any particular worries, please do not hesitate to ask your audiologist or doctor. What is conductive hearing loss? Your ear is made up of three sections; the outer, middle and inner ear. Conductive hearing loss occurs when there is an obstruction (blockage) in the outer or middle ear that prevents sound from passing through them properly. This type of hearing loss can be treated or managed depending on what is causing the conductive impairment. What causes conductive hearing loss? There are many reasons a person could have conductive hearing loss: a foreign body in the ear canal/s impacted wax in the ear canal/s fluid in the middle ear due to colds or allergies (otitis media) an ear infection a hole in the ear drum/s (perforation) eustachian tube dysfunction abnormal growth in the ear canal/s abnormal formation of the outer ear (atresia/microtia) damage to the bones in the middle ear failed surgery What happens next? In most cases, conductive hearing loss can be successfully managed by treating the underlying cause. For example, blockage in the ear canal due to wax can be treated by having wax removed. If there is fluid in the ear, antibiotics may be prescribed by your GP to help resolve the infection. In some cases, the use of a hearing aid or hearing aids can be beneficial to help with the transmission of sound. If a conventional hearing aid fails to provide any benefit, the use of a bone anchored hearing aid (BAHA) is an option. -

Hearing Loss Descriptions
DESCRIPTIONS OF HEARING LOSS As your child participates in hearing testing, you will hear hearing loss described in different ways. This section explains all about hearing loss. TYPE OF HEARING LOSS How Do We Hear? The Ear is made up of three parts: Outer Ear The outer ear is made up of three parts; the part we see on the sides of our heads (pinna), the ear canal (external auditory canal), and the eardrum (tympanic membrane). Sound travels through the ear canal and makes the eardrum move. Middle Ear The middle ear is made up of the eardrum and three small bones (ossicles). When the eardrum moves, the three middle ear bones vibrate. This vibration creates movement of fluid in the inner ear. Inner Ear The inner ear is made up of the snail shaped organ for hearing (cochlea), and the hearing (Auditory) nerve that go to the brain The movement of fluid causes the inner ear to send nerve signals to the brain. Once the brain receives the message, it identifies that message as sound. What are the Different Types of Hearing Loss? Conductive Hearing Loss This type of hearing loss is caused by a problem in the outer or middle ear. This means that sound is having difficulty traveling to the inner ear. Children with conductive hearing loss usually cannot hear faint sounds. Some causes of conductive hearing loss are fluid in the middle ear, wax in the ear canal or a hole in the eardrum. Most types of conductive hearing loss can be treated with medicine or surgery.