Deafness and Hereditary Hearing Loss Overview - Genereviews™ - NCBI Bookshelf Page 1 of 25
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Age-Related Hearing Loss
U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES ∙ National Institutes of Health NIDCD Fact Sheet | Hearing and Balance Age-Related Hearing Loss What is age-related hearing loss? The auditory system Age-related hearing loss (presbycusis) is the loss of hearing that gradually occurs in most of us as we grow older. It is one of the most common conditions affecting older and elderly adults. Approximately one in three people in the United States between the ages of 65 and 74 has hearing loss, and nearly half of those older than 75 have difficulty hearing. Having trouble hearing can make it hard to understand and follow a doctor’s advice, respond to warnings, and hear phones, doorbells, and smoke alarms. Hearing loss can also make it hard to enjoy talking with family and friends, leading to feelings of isolation. Age-related hearing loss most often occurs in both ears, affecting them equally. Because the loss is gradual, if you have age-related hearing loss you Credit: NIH Medical Arts may not realize that you’ve lost some of your ability to hear. How do we hear? There are many causes of age-related hearing Hearing depends on a series of events that change loss. Most commonly, it arises from changes in sound waves in the air into electrical signals. Your the inner ear as we age, but it can also result auditory nerve then carries these signals to your from changes in the middle ear, or from complex brain through a complex series of steps. changes along the nerve pathways from the ear 1. -

Endoplasmic Reticulum Stress As Target for Treatment of Hearing Loss
REVIEW ARTICLE Endoplasmic reticulum stress as target for treatment of hearing loss Yanfei WANG, Zhigang XU* Shandong Provincial Key Laboratory of Animal Cell and Developmental Biology, School of Life Sciences, Shandong University, Qingdao, Shandong 266237, China *Correspondence: [email protected] https://doi.org/10.37175/stemedicine.v1i3.21 ABSTRACT The endoplasmic reticulum (ER) plays pivotal roles in coordinating protein biosynthesis and processing. Under ER stress, when excessive misfolded or unfolded proteins are accumulated in the ER, the unfolded protein response (UPR) is activated. The UPR blocks global protein synthesis while activates chaperone expression, eventually leading to the alleviation of ER stress. However, prolonged UPR induces cell death. ER stress has been associated with various types of diseases. Recently, increasing evidences suggest that ER stress and UPR are also involved in hearing loss. In the present review, we will discuss the role of ER stress in hereditary hearing loss as well as acquired hearing loss. Moreover, we will discuss the emerging ER stress-based treatment of hearing loss. Further investigations are warranted to understand the mechanisms in detail how ER stress contributes to hearing loss, which will help us develop better ER stress-related treatments. Keywords: ER stress · Unfolded protein response (UPR) · Hearing loss · Inner ear · Cochlea 1. Introduction far, which are mediated by ER stress sensors that reside The endoplasmic reticulum (ER) is a highly dynamic on the ER membranes, namely the inositol-requiring organelle in eukaryotic cells, playing important roles in enzyme 1α (IRE1α), the PKR-like ER kinase (PERK), protein synthesis, processing, folding, and transportation, and the activating transcription factor 6α (ATF6α) as well as lipid synthesis and calcium homeostasis. -

Mutations in the WFS1 Gene Are a Frequent Cause of Autosomal Dominant Nonsyndromic Low-Frequency Hearing Loss in Japanese
J Hum Genet (2007) 52:510–515 DOI 10.1007/s10038-007-0144-3 ORIGINAL ARTICLE Mutations in the WFS1 gene are a frequent cause of autosomal dominant nonsyndromic low-frequency hearing loss in Japanese Hisakuni Fukuoka Æ Yukihiko Kanda Æ Shuji Ohta Æ Shin-ichi Usami Received: 14 January 2007 / Accepted: 27 March 2007 / Published online: 11 May 2007 Ó The Japan Society of Human Genetics and Springer 2007 Abstract Mutations in WFS1 are reported to be respon- sites are likely to be mutational hot spots. All three families sible for two conditions with distinct phenotypes; DFNA6/ with WFS1 mutations in this study showed a similar phe- 14/38 and autosomal recessive Wolfram syndrome. They notype, LFSNHL, as in previous reports. In this study, one- differ in their associated symptoms and inheritance mode, third (three out of nine) autosomal dominant LFSNHL and although their most common clinical symptom is families had mutations in the WFS1 gene, indicating that in hearing loss, it is of different types. While DNFA6/14/38 is non-syndromic hearing loss WFS1 is restrictively and characterized by low frequency sensorineural hearing loss commonly found within autosomal dominant LFSNHL (LFSNHL), in contrast, Wolfram syndrome is associated families. with various hearing severities ranging from normal to profound hearing loss that is dissimilar to LFSNHL (Pen- Keywords WSF1 Á Low-frequency hearing loss Á nings et al. 2002). To confirm whether within non-syn- DFNA6/14/38 dromic hearing loss patients WFS1 mutations are found restrictively in patients with LFSNHL and to summarize the mutation spectrum of WFS1 found in Japanese, we Introduction screened 206 Japanese autosomal dominant and 64 auto- somal recessive (sporadic) non-syndromic hearing loss WFS1 is a gene encoding an 890 amino-acid glycoprotein probands with various severities of hearing loss. -

Fumihiko Urano: Wolfram Syndrome: Diagnosis, Management, And
Curr Diab Rep (2016) 16:6 DOI 10.1007/s11892-015-0702-6 OTHER FORMS OF DIABETES (JJ NOLAN, SECTION EDITOR) Wolfram Syndrome: Diagnosis, Management, and Treatment Fumihiko Urano1,2 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Wolfram syndrome is a rare genetic disorder char- diabetes insipidus, optic nerve atrophy, hearing loss, and acterized by juvenile-onset diabetes mellitus, diabetes neurodegeneration. It was first reported in 1938 by Wol- insipidus, optic nerve atrophy, hearing loss, and neurodegen- fram and Wagener who found four of eight siblings with eration. Although there are currently no effective treatments juvenile diabetes mellitus and optic nerve atrophy [1]. that can delay or reverse the progression of Wolfram syn- Wolfram syndrome is considered a rare disease and esti- drome, the use of careful clinical monitoring and supportive mated to afflict about 1 in 160,000–770,000 [2, 3]. In care can help relieve the suffering of patients and improve 1995, Barrett, Bundey, and Macleod described detailed their quality of life. The prognosis of this syndrome is current- clinical features of 45 patients with Wolfram syndrome ly poor, and many patients die prematurely with severe neu- and determined the best available diagnostic criteria for rological disabilities, raising the urgency for developing novel the disease [3]. According to the draft International Clas- treatments for Wolfram syndrome. In this article, we describe sification of Diseases (ICD-11), Wolfram Syndrome is natural history and etiology, provide recommendations for categorized as a rare specified diabetes mellitus (subcate- diagnosis and clinical management, and introduce new treat- gory 5A16.1, Wolfram Syndrome). -

Benefits of Exome Sequencing in Children with Suspected Isolated
G C A T T A C G G C A T genes Article Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss Roxane Van Heurck 1, Maria Teresa Carminho-Rodrigues 1, Emmanuelle Ranza 1, Caterina Stafuzza 2, Lina Quteineh 1, Corinne Gehrig 1, Eva Hammar 1, Michel Guipponi 1, Marc Abramowicz 1, Pascal Senn 2 , Nils Guinand 2, Helene Cao-Van 2 and Ariane Paoloni-Giacobino 1,* 1 Division of Genetic Medicine, Geneva University Hospitals, 1205 Geneva, Switzerland; [email protected] (R.V.H.); [email protected] (M.T.C.-R.); [email protected] (E.R.); [email protected] (L.Q.); [email protected] (C.G.); [email protected] (E.H.); [email protected] (M.G.); [email protected] (M.A.) 2 Ear-Nose-Throat/Head and Neck Surgery Division, Geneva University Hospitals, 1205 Geneva, Switzerland; [email protected] (C.S.); [email protected] (P.S.); [email protected] (N.G.); [email protected] (H.C.-V.) * Correspondence: [email protected] Abstract: Purpose: Hearing loss is characterized by an extensive genetic heterogeneity and remains a common disorder in children. Molecular diagnosis is of particular benefit in children, and permits the early identification of clinically-unrecognized hearing loss syndromes, which permits effective clinical management and follow-up, including genetic counselling. Methods: We performed whole-exome Citation: Van Heurck, R.; sequencing with the analysis of a panel of 189 genes associated with hearing loss in a prospective Carminho-Rodrigues, M.T.; Ranza, E.; cohort of 61 children and 9 adults presenting mainly with isolated hearing loss. -

The ENT Manifestation of Wolfram Syndrome (DIDMOAD): a Case Report
Journal of Clinical Science & Translational Medicine MEDWIN PUBLISHERS Committed to Create Value for Researchers The ENT Manifestation of Wolfram Syndrome (DIDMOAD): A Case Report Gliti MA1,3*, Allouche I1,3, Razika B2,3, Anas BM2,3 and Houssyni LE2,3 Case Report 1Department of Otorhinolaryngology, Head and Neck Surgery, Ibn Sina University Hospital, Volume 3 Issue 1 Morocco Received Date: May 15, 2021 2Department of Otorhinolaryngology, Head and Neck Surgery, Ibn Sina University Hospital, Published Date: June 23, 2021 Morocco 3Faculty of Medicine and Pharmacy of Rabat, Mohammed V University, Morocco *Corresponding author: Mohamed Ali Gliti, Department of Otorhinolaryngology, Head and Neck Surgery, Ibn Sina University Hospital, Rabat, Morocco, Tel: 0633725750; Email: [email protected] Abstract Objective: Describe the clinical and therapeutic aspects of WOLFRAM syndrome (DIDMOAD) presenting with deafness. Materials and Methods: We report the case of a 21-year-old man who presented with a WOLFRAM syndrome associated with a tympanic perforation. Clinical Case: This is a 21-year-old R.Y from a consanguineous marriage (first cousin 1st degree) Due to the association syndrome. of symptoms (type 1 diabetes, urinary and ophthalmologic signs), genetic counseling was sought to confirm WOLFRAM Conclusion: vigilant in referring patients with hearing loss for an ophthalmic examination. Since sensorineural hearing loss can be the first symptom of SW, audiologists, and otolaryngologists should be Keywords: WOLFRAM Syndrome; Sensorineural Hearing Loss; Tympanic Perforation; Tympanoplasty Abbreviations: SW: Wolfram Syndrome. feature. For this reason, the same Wolfram syndrome (SW) Introduction (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness)is defined in with the literaturethe term [2-7].Wolfram It is syndromea recessive DIDMOAD inherited disease, the pathogenesis of which is still poorly understood Wagener, a clinical feature characterized by diabetes [8-10]. -

Dissertationes Medicinae Universitatis Tartuensis 178
DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 178 DISSERTATIONES MEDICINAE UNIVERSITATIS TARTUENSIS 178 RITA TEEK The genetic causes of early onset hearing loss in Estonian children Department of Paediatrics, University of Tartu, Tartu, Estonia Dissertation is accepted for commencement of the degree of Doctor of Medical Sciences on September 22, 2010 by the Council of the Faculty of Medicine, University of Tartu, Estonia. Supervisors: Professor Katrin Õunap, MD, PhD, Department of Paediatrics, University of Tartu, Tartu, Estonia The Late Professor Mart Kull, MD, PhD, Department of Oto-Rhino-Laryngology, University of Tartu, Tartu, Estonia (2005–2008) Reviewers: Assistant Professor Gunnar Tasa, MD, PhD, Department of General and Molecular Pathology, University of Tartu, Tartu, Estonia Assistant Professor Oivi Uibo, MD, PhD, Department of Paediatrics, University of Tartu, Tartu, Estonia Opponent: Professor Lisbeth Tranebjærg, MD, PhD, Department of Audiology, H:S Bispebjerg Hospital, and Wilhelm Johannsen Centre of Functional Genomics Institute of Cellular and Molecular Medicine, ICMM, University of Copenhagen, The Panum Institute, Denmark Commencement: November 24, 2010 ISSN 1024–395x ISBN 978–9949–19–478–0 (trükis) ISBN 978–9949–19–479–7 (PDF) Autoriõigus: Rita Teek, 2010 Tartu Ülikooli Kirjastus www.tyk.ee Tellimuse nr. 570 To my patients and their families CONTENTS LIST OF ORIGINAL PUBLICATIONS ...................................................... 9 ABBREVIATIONS OF HEARING LOSS STUDY GROUPS AND PATIENTS .................................................................................................... -
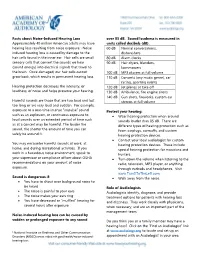
Facts About Noise-Induced Hearing Loss Over 85 Db
Facts about Noise-Induced Hearing Loss over 85 dB. Sound loudness is measured in Approximately 40 million American adults may have units called decibels (dB). hearing loss resulting from noise exposure.1 Noise- 60 dB Normal conversations, induced hearing loss is caused by damage to the dishwashers hair cells found in the inner ear. Hair cells are small 80 dB Alarm clocks sensory cells that convert the sounds we hear 90 dB Hair dryers, blenders, (sound energy) into electrical signals that travel to lawnmowers the brain. Once damaged, our hair cells cannot 100 dB MP3 players at full volume grow back, which results in permanent hearing loss. 110 dB Concerts (any music genre), car racing, sporting events Hearing protection decreases the intensity, or 120 dB Jet planes at take off loudness, of noise and helps preserve your hearing. 130 dB Ambulance, fire engine sirens 140 dB Gun shots, fireworks, custom car Harmful sounds are those that are too loud and last stereos at full volume too long or are very loud and sudden. For example, exposure to a one-time intense “impulse” sound Protect your hearing: such as an explosion, or continuous exposure to • Wear hearing protection when around loud sounds over an extended period of time such sounds louder than 85 dB. There are as at a concert may be harmful. The louder the different types of hearing protection such as sound, the shorter the amount of time you can foam earplugs, earmuffs, and custom safely be around it. hearing protection devices • Contact your local audiologist for custom You may encounter harmful sounds at work, at hearing protection devices. -

The Epidemiology of Deafness
Downloaded from http://perspectivesinmedicine.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press The Epidemiology of Deafness Abraham M. Sheffield1 and Richard J.H. Smith2,3,4,5 1Department of Otolaryngology, Head and Neck Surgery, University of Iowa, Iowa City, Iowa 52242 2Molecular Otolaryngology and Renal Research Laboratories (MORL), Department of Otolaryngology, University of Iowa, Iowa City, Iowa 52242 3Department of Molecular Physiology & Biophysics, University of Iowa, Iowa City, Iowa 52242 4Department of Pediatrics, University of Iowa, Iowa City, Iowa 52242 5Department of Internal Medicine, University of Iowa, Iowa City, Iowa 52242 Correspondence: [email protected] Hearing loss is the most common sensory deficit worldwide. It affects ∼5% of the world population, impacts people of all ages, and exacts a significant personal and societal cost. This review presents epidemiological data on hearing loss. We discuss hereditary hearing loss, complex hearing loss with genetic and environmental factors, and hearing loss that is more clearly related to environment. We also discuss the disparity in hearing loss across the world, with more economically developed countries having overall lower rates of hearing loss compared with developing countries, and the opportunity to improve diagnosis, preven- tion, and treatment of this disorder. earing loss is the most common sensory refer to people with mild-to-moderate (and Hdeficit worldwide, affecting more than half sometimes severe) hearing loss, whereas the a billion people (Smith et al. 2005; Wilson et al. term “deaf” (lower case “d”) is more commonly 2017). Normal hearing is defined as having hear- reserved for those with severe or profound hear- ing thresholds of ≤25 dB in both ears. -

Hearing Loss
Randal W. Swenson, M.D. Joshua G. Yorgason, M.D. David K. Palmer, M.D. Wesley R. Brown, M.D. John E. Butler, M.D. Nancy J. Stevenson, PA-C Justin D. Gull, M.D. ENT SPECIALISTS Kristin G. Hoopes, PA-C www.entslc.com Hearing Loss Approximately one in ten persons in the United may result from blockage of the ear canal (wax), States has some degree of hearing loss. Hearing is from a perforation (hole) in the ear drum, or from measured in decibels (dB), and a hearing level of 0- infection or disease of any of the three middle ear 25 dB is considered normal hearing. Your level is: bones. With a conductive loss only, the patient will never go deaf, but will always be able to hear, either Right ear _______ dB Left ear _______dB with reconstructive ear surgery or by use of a properly fitted hearing aid. Some patients who are Hearing Severity / % Loss not candidates for surgery, may benefit from a new 25 dB (normal).….0% 65dB(Severe)……...60% technology, the Baha (bone-anchored hearing aid). 35 dB (mild)……..15% 75dB(Severe)……...75% When there is a problem with the inner ear or 45 dB (moderate)..30% >85dB (Profound)..>90% nerve of hearing, a sensori-neural hearing loss occurs. This is most commonly from normal aging, Normal speech discrimination is 88-100%. Yours is: is usually worse in high frequencies, and can progress to total deafness. Noise exposure is another Right ear _______ % Left ear_______% common cause of high frequency hearing loss. Patients with sensori-neural hearing loss usually complain of difficulty hearing in loud environments. -

A New Age in the Genetics of Deafness
, September/October 1999. Vol. 1 . No. 6 invited review/cme article A new age in the genetics of deafness Heidi L. Rehtu, MMSC' ot~dCytltllia C. Morton, PAD' Over the last several years, an understanding of the genetics SYNDROMIC AND NONSYNDROMIC DEAFNESS of hearing and deafness has grown exponentially. This progress , The prevalence of severe to profound bilateral congenital has been fueled by fast-paced developments in gene mapping hearing loss is estimated at 1 in 1000 births? When milder I and discovery, leading to the recent cloning and characteriza- forms of permanent hearing impairment at birth are included, tion of many genes critical in the biology of hearing. Among this estimate increases four-fold. Congenital deafness refers to the most significant advances, especially from a clinical per- deafness present at birth, though the cause of such hearing spective, is the discovery that up to 50% of nonsyndromic re- impairment may be genetic (hereditary) or environmental (ac- cessive deafness is caused by mutations in the GIB2 gene, which quired). Various studies estimate that approximately one-half encodes the connexin 26 protein (Cx26).I.' The clinical use of of the cases of congenital deafness are due to genetic factor^,^ screening for mutations in Cx26 and other genes involved in and these factors can be further subdivided by mode of inher- hearing impairment is still a new concept and not widely avail- itance. Approximately 77% of cases of hereditary deafness are able. However, the eventual availability of an array of genetic autosomal recessive, 22% are autosomal dominant, and 1% are tests is likely to have a major impact on the medical decisions X-linked.-l In addition, a small fraction of hereditary deafness made by hearing-impaired patients, their families, and their (< 1%) represents those families with mitochondria1 inheri- physicians. -

The Pathological Consequences of Impaired Genome Integrity in Humans; Disorders of the DNA Replication Machinery
The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery Article (Accepted Version) O'Driscoll, Mark (2017) The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery. Journal of Pathology, 241 (2). pp. 192-207. ISSN 0022-3417 This version is available from Sussex Research Online: http://sro.sussex.ac.uk/id/eprint/65956/ This document is made available in accordance with publisher policies and may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the URL above for details on accessing the published version. Copyright and reuse: Sussex Research Online is a digital repository of the research output of the University. Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. To the extent reasonable and practicable, the material made available in SRO has been checked for eligibility before being made available. Copies of full text items generally can be reproduced, displayed or performed and given to third parties in any format or medium for personal research or study, educational, or not-for-profit purposes without prior permission or charge, provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. http://sro.sussex.ac.uk The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery.