Infectious RNA Transcripts from Full-Length Dengue Virus Type 2 Cdna Clones Made in Yeast
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bpuami: a Novel Saci Neoschizomer from Bacillus Pumilus Discovered in an Isolate from Amazon Basin, Recognizing 5'-Gag↓Ctc-3'
Brazilian Journal of Microbiology (2006) 37:96-100 ISSN 1517-8382 BPUAMI: A NOVEL SACI NEOSCHIZOMER FROM BACILLUS PUMILUS DISCOVERED IN AN ISOLATE FROM AMAZON BASIN, RECOGNIZING 5'-GAG↓CTC-3' Jocelei M. Chies1,2,*; Ana C. de O. Dias1; Hélio M. M. Maia3; Spartaco Astolfi-Filho2,3 1Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, RS, Brasil; 2Curso de Pós-Graduação em Biologia Molecular, Universidade de Brasília, Brasília, DF, Brasil; 3Centro de Apoio Multidisciplinar, Universidade Federal do Amazonas, Manaus, AM, Brasil Submitted: June 27, 2005; Returned to authors for corrections: November 16, 2005; Approved: January 12, 2006 ABSTRACT A strain of Bacillus pumilus was isolated and identified from water samples collected from a small affluent of the Amazon River. Type II restriction endonuclease activity was detected in these bacteria. The enzyme was purified and the molecular weight of the native protein estimated by gel filtration and SDS-PAGE. The optimum pH, temperature and salt requirements were determined. Quality control assays showed the complete absence of “nonspecific nucleases.” Restriction cleavage analysis and DNA sequencing of restriction fragments allowed the unequivocal demonstration of 5´GAG↓CTC3´ as the recognition sequence. This enzyme was named BpuAmI and is apparently a neoschizomer of the prototype restriction endonuclease SacI. This is the first report of an isoschizomer and/or neoschizomer of the prototype SacI identified in the genus Bacillus. Key words: type II restriction endonuclease, BpuAmI, SacI, neoschizomers, Bacillus pumilus INTRODUCTION thousands of taxonomically diverse bacteria for enzymes with new characteristics. However, to our knowledge, there has Restriction endonucleases are enzymes which recognize not been any report to date of an isoschizomer and/or short DNA sequences and cleave DNA in both strands. -
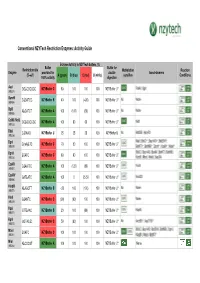
Conventional Nzytech Restriction Enzymes: Activity Guide
Conventional NZYTech Restriction Enzymes: Activity Guide Enzyme Activity in NZYTech buffers (%) Buffer Buffer for Restriction site Methylation Reaction Enzyme provided for double Isoschizomers (5’ →→→3’) A (green) B (blue) C (red) U (white) sensitive Conditions 100% activity digestion AscI GG ↓↓↓CGCGCC NZYBuffer C 80 100 100 100 NZYBuffer U * PalAI, SgsI (MB231) BamHI G↓↓↓GATCC NZYBuffer B 40 100 (<20) 100 NZYBuffer U * No None (MB064) BglII A↓↓↓GATCT NZYBuffer A 100 (100) (60) 100 NZYBuffer U * No None (MB065) CciNI (NotI) GC ↓↓↓GGCCGC NZYBuffer A 100 80 60 100 NZYBuffer U * NotI (MB153) DdeI C↓↓↓TNAG NZYBuffer U 25 25 25 100 NZYBuffer U No BstDEI, HpyF3I (MB236) MalI, BfuCI ♦♦♦, Bsp143I ♦♦♦, BstENII ♦♦♦, DpnI G(mA)↓↓↓TC NZYBuffer C 70 50 100 100 NZYBuffer U * (MB078) BstKTI ♦♦♦, BstMBI ♦♦♦, DpnII ♦♦♦, Kzo9I ♦♦♦, NdeII ♦♦♦ BfuCI, Bsp143I, BssMI, BstKTI, BstMBI, DpnII ↓↓↓GATC NZYBuffer C 80 80 100 100 NZYBuffer U * (MB233) Kzo9I, MboI, NdeII, Sau3AI EcoRI G↓↓↓AATTC NZYBuffer A 100 (120) (80) 100 NZYBuffer U * FunII (MB067) EcoRV GAT ↓↓↓ATC NZYBuffer A 100 0 25-50 100 NZYBuffer U * Eco32I (MB068) HindIII A↓↓↓AGCTT NZYBuffer B <20 100 (100) 100 NZYBuffer U * No None (MB070) HinfI G↓↓↓ANTC NZYBuffer C (90) (90) 100 100 NZYBuffer U * None (MB239) HpaI GTT ↓↓↓AAC NZYBuffer B 20 100 (80) 100 NZYBuffer U * KspAI (MB071) KpnI GGTAC ↓↓↓C NZYBuffer C 50 (80) 100 100 NZYBuffer U * No Acc65I ♦♦♦, Asp718I ♦♦♦ (MB072) BfuCI, Bsp143I, BstENII, BstMBI, DpnII, MboI ↓↓↓GATC NZYBuffer C 100 100 100 100 NZYBuffer U * (MB241) Kzo9II, NdeII, BstKTI ♦♦♦ MluI -

Curriculum Vitae SIR RICHARD JOHN ROBERTS ADDRESS PERSONAL
Curriculum Vitae SIR RICHARD JOHN ROBERTS ADDRESS New England Biolabs 240 County Road, Ipswich, MA 02138 USA Email: [email protected] Telephone: (978) 380-7405 / Fax: (978) 380-7406 PERSONAL Born on September 6, 1943, Derby, England EDUCATION 1962-1965 University of Sheffield, Sheffield, England B.Sc. in Chemistry 1966-1968 University of Sheffield, Sheffield, England Ph.D. in Organic Chemistry POSITIONS 2005- Chief Scientific Officer, New England Biolabs 1992-2005 Research Director, New England Biolabs 1986-92 Assistant Director for Research, Cold Spring Harbor Laboratory 1972-86 Senior Staff Investigator, Cold Spring Harbor Laboratory 1971-1972 Research Associate in Biochemistry, Harvard University 1969-1970 Research Fellow, Harvard University OUTSIDE ACTIVITIES 1974-1992 Consultant and Chairman of Scientific Advisory Board New England Biolabs 1977-1985 Scientific Advisory Board, Genex Corp. 1977-1987 Editorial Board: Nucleic Acids Research 1979-1984 Editorial Board: Journal of Biological Chemistry 1982-1989 Member: National Advisory Committee of GENBANK 1984-1986 Member: National Advisory Committee of BIONET 1985-1988 Panel member: NIH Study Section in Biochemistry. 1985-2002 Editorial Board: Bioinformatics (formerly CABIOS) 1987-1990 Chairman: National Advisory Committee of BIONET 1987-2009 Senior Executive Editor: Nucleic Acids Research 1990-1992 Panel member: NCI Cancer Centers Support Grant Review Committee 1993-1995 Panel member: NLM Study Section/Comp. Biol. 1994-2000 Scientific Advisory Board, Molecular Tool 1994- Patron of the Oxford International Biomedical Center 1996-1998 Visiting Professor, University of Bath, UK. 1996-2000 Chairman, NCI Board of Scientific Counselors 1996-1999 Scientific Advisory Board, Oxford Molecular Group 1997-2001 Editorial Board: Current Opinion Chem. Biol. -

Restriction Enzymes Are Molecular Scissors
Molecular Scissors Restriction enzymes are molecular scissors DR. ABUL HASAN SARDAR Assistant Professor and Head Department of Microbiology Sarsuna College RESTRICTION ENZYMES • A restriction enzyme (or restriction endonuclease) is an enzyme that cuts double- stranded or single stranded DNA at specific recognition nucleotide sequences known as restriction sites. Property of restriction enzymes • They that link adjacent nucleotides in DNA molecules. HOW RESTRICTION ENZYMES WORKS? • Restriction enzymes recognize a specific sequence of nucleotides, and produce a double-stranded cut in the DNA, these cuts are of two types: • BLUNT ENDS. • STICKY ENDS. Blunt end Sticky end BLUNT ENDS • These blunt ended fragments can be joined to any other DNA fragment with blunt ends. • Enzymes useful for certain types of DNA cloning experiments “STICKY ENDS” ARE USEFUL DNA fragments with complimentary sticky ends can be combined to create new molecules which allows the creation and manipulation of DNA sequences from different sources. • While recognition sequences vary widely , with lengths between 4 and 8 nucleotides, many of them are palindromic. PALINDROMES IN DNA SEQUENCES Genetic palindromes are similar to verbal palindromes. A palindromic sequence in DNA is one in which the 5’ to 3’ base pair sequence is identical on both strands (the 5’ and 3’ ends refers to the chemical structure of the DNA). PALINDROME SEQUENCES • The mirror like palindrome in which the same forward and backwards are on a single strand of DNA strand, as in GTAATG • The Inverted repeat palindromes is also a sequence that reads the same forward and backwards, but the forward and backward sequences are found in complementary DNA strands (GTATAC being complementary to CATATG) • Inverted repeat palindromes are more common and have greater biological importance than mirror- like palindromes. -

Review Type II Restriction Endonucleases
CMLS, Cell. Mol. Life Sci. 62 (2005) 685–707 1420-682X/05/060685-23 DOI 10.1007/s00018-004-4513-1 CMLS Cellular and Molecular Life Sciences © Birkhäuser Verlag, Basel, 2005 Review Type II restriction endonucleases: structure and mechanism A. Pingouda,*, M. Fuxreiterb, V.Pingouda and W.Wendea a Institut für Biochemie, Justus-Liebig-Universität, Heinrich-Buff-Ring 58, 35392 Giessen (Germany), Fax: +49 641 9935409, e-mail: [email protected] b Institute of Enzymology, Biological Research Centre, Hungarian Academy of Sciences, Karolina ut 29, 1113 Budapest (Hungary) Received 15 November 2004; accepted 9 December 2004 Abstract. Type II restriction endonucleases are compo- few restriction endonucleases discovered thus far do not nents of restriction modification systems that protect belong to the PD…D/ExK family of enzymes, but rather bacteria and archaea against invading foreign DNA. Most have active sites typical of other endonuclease families. are homodimeric or tetrameric enzymes that cleave DNA The present review deals with new developments in the at defined sites of 4–8 bp in length and require Mg2+ ions field of Type II restriction endonucleases. One of the for catalysis. They differ in the details of the recognition more interesting aspects is the increasing awareness of process and the mode of cleavage, indicators that these the diversity of Type II restriction enzymes. Nevertheless, enzymes are more diverse than originally thought. Still, structural studies summarized herein deal with the more most of them have a similar structural core and seem to common subtypes. A major emphasis of this review will share a common mechanism of DNA cleavage, suggest- be on target site location and the mechanism of catalysis, ing that they evolved from a common ancestor. -

The Mmei Family: Type II Restriction–Modification Enzymes That Employ Single-Strand Modification for Host Protection Richard D
5208–5221 Nucleic Acids Research, 2009, Vol. 37, No. 15 Published online 3 July 2009 doi:10.1093/nar/gkp534 The MmeI family: type II restriction–modification enzymes that employ single-strand modification for host protection Richard D. Morgan*, Elizabeth A. Dwinell, Tanya K. Bhatia, Elizabeth M. Lang and Yvette A. Luyten New England Biolabs, Inc. 240 County Road Ipswich, MA 01938 USA Received March 18, 2009; Revised and Accepted June 8, 2009 ABSTRACT material between ‘self’ and ‘others’ within microbial popu- lations (2). As such they represent fertile ground for the The type II restriction endonucleases form one of study of protein evolution, particularly the evolution the largest families of biochemically-characterized of those protein–DNA interactions that confer sequence proteins. These endonucleases typically share little specificity to these enzymes. sequence similarity, except among isoschizomers A restriction system must differentiate between ‘self’ that recognize the same sequence. MmeI is an unu- and ‘foreign’ DNA in order to cut the identifiably foreign sual type II restriction endonuclease that combines DNA but not host DNA. This discrimination is based endonuclease and methyltransferase activities in a upon modification of the DNA, usually in the form of single polypeptide. MmeI cuts DNA 20 bases from methylation of a base in each strand within the discrete its recognition sequence and modifies just one DNA DNA sequence recognized by the endonuclease. Usually, strand for host protection. Using MmeI as query we hemi-methylated DNA is not cut by the type II endonu- have identified numerous putative genes highly sim- cleases, ensuring that immediately following replication the hemi-methylated daughter chromosomes remain pro- ilar to MmeI in database sequences. -

Assignment 1 Part I
1 Assignment 1 Note: Part I of the assignment is to be submitted through Blackboard (See instructions on Blackboard). Parts II, III and IV are to be submitted as a hard copy during your lab session. Part I Dilutions and Concentrations When doing your calculations, do not round off your intermediate numbers. Only round off the final answer. Your answers must be submitted to two significant figures after the decimal. For example 2.00, 0.020, or 0.0020. It is strongly recommended that you submit your answers using the web browser Firefox. You will be allowed two submissions! Use the following information to answer questions 1-6 You prepare a solution by adding the following ingredients in the order indicated: 600 mL H2O, 125 mL 1.6 M LiCl, 50 mL 20 % (m/v) MgCl2, and 25 mL 10g/L NaCl. The properties of each ingredient are as follows: LiCl: MW 200g/mole, density 1.2g/mL, density of 1.6 M soln. 1.05g/mL MgCl2: MW 150g/mole, density 1.3g/mL, density of 20% soln. 1.1g/mL NaCl: MW 35g/mole, density 1.15g/mL, density of 10g/L soln. 1.03g/mL Final solution: density: 1.25g/mL 1. What is the final molarity of MgCl2 in the solution? (0.083M) 2. What is the volume in milliliters of one part? (25 mL) 3. What is the percentage (m/m) of NaCl in the final solution? (0.025%) 4. What is the percentage (m/v) of LiCl in the final solution? (5%) 5. What is the number of parts of solvent in the final solution? (24 parts) 6. -

R.Sspd5i Is a Neoschizomer of Hphi Producing Blunt End DNA Fragments
FEBS Letters 448 (1999) 38^40 FEBS 21798 R.SspD5I is a neoschizomer of HphI producing blunt end DNA fragments Lyudmila Zheleznayaa, Sergey Shiryaevb, Elena Zheleznyakovab, Nadezhda Matvienkoc, Nicholas Matvienkob;* aInstitute of Theoretical and Applied Biophysics, RAS, 142292 Pushchino, Russia bInstitute of Protein Research, RAS, 142292 Pushchino, Russia cBranch of the Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, RAS, 142292 Pushchino, Russia Received 18 February 1999 reaction bu¡er (10 mM Tris-HCl, pH 7.4, 100 Wg/ml albumin, 10 mM Abstract The strain Staphylococcus species D5 produces a MgCl2) were 10, 50 and 100 mM for LRB, MRB and HRB, respec- restriction enzyme. It is the neoschizomer of HphI endonuclease, tively. The pH values of the reaction bu¡er were 7.0, 7.4, 8.0. Incu- which cleaves DNA at a distance of eight nucleotides from the bation temperatures were 28, 37 and 48³C. recognition sequences producing blunt end DNA fragments: 5P- GGTGA8Ns-3P and 3P-CCACT8Nu-5P. 2.3. Determination of DNA cleavage points z 1999 Federation of European Biochemical Societies. Cleavage points on DNA were determined by the elongated primer method [6]. As a template, we used for the ¢rst time the recombinant phage DNAs specially constructed for this study (M13tg130: Key words: Site-speci¢c endonuclease; Neoschizomer; VCIHindIII and M13tg131: VCIHindIII) containing the small frag- Restriction ment of VCI857S7 DNA with coordinates 37459^37584 inserted in opposite orientations. These phage DNAs contained the recognition site for the SspD5I endonuclease near the universal primer. To make DNA recombinants containing multiple recognition sites 1. Introduction for the endonuclease SspD5I in di¡erent surrounding nucleotide se- quences, we used a synthetic oligonucleotide: 5P-AGATCT11NT- Site-speci¢c endonucleases are in the forefront of the en- CACC3NGGATCC-3P, which contains the recognition site for zymes used in genetic engineering. -

Restriction-Enzymes
Methods for cutting DNA Chemical methods Mechanical shearing in a blender Restriction Endonuclease digestion sonication Restriction and Modification systems: reported in several bacteria s Function: Restriction systems checks the presence of foreign DNA and destroys it. s recognize specific sequences in the incoming DNA and breaks into fragments. s Cutting is either at specific sites or random. Discovery s Bacteriophages: have a single host they can infect, or a small number of related bacteria: which is the ‘host range’ s This effect is seen in efficiency of plating, i.e. the number of plaques formed in plating tests gets reduced. s Observation: In the 50’s and 60’s occasionally a virus could become active in a new strain but would no longer be effective in the original host Discovery s This change in host range seemed to be associated with specific bases in the viral DNA being methylated: s This is called modification : a change conferred by the host Methylases s 1969 Werner Arber and Stewart Linn discovered that most bacteria contained a class of enzymes which modify the bases in DNA termed methylases. s These enzymes recognize a specific sequence of DNA and add a methyl group onto certain bases, most often an adenine or cytosine. s The presence of these methyl groups on the viral DNA seemed to be the cause of their ability to grow in some strains of bacteria, but not others. methylation effect: s This ability to grow was because the methyl groups were protecting the viral DNA from an unusual type of enzyme. -

(I /1 Alan Grodzinsky
Analysis of the Structural Changes Caused by Positive DNA Supercoiling by- Marita Christine Barth B.S. Bioresource Research Oregon State University, 1998 Submitted to the Division of Biological Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Macromolecular Biochemistry and Biophysics at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2007 C 2007 Massachusetts Institute of Technology. All rights reserved. Signature of Author: Division of Biological Engineering January 12, 2007 Certified by: (I Peter C. Dedon Professor of Toxicology and Biological Engineering Thesis Supervisor Accepted by: L". % SLr/ .,: Alan Grodzinsky Irof's r of Biological Engineering rMASSACK·KIEMEWIJf nNte OF TECHNOLOGY /1 Chairman, Co ittee for Graduate Students AUG 0 2 2007I LIBRARIES ARQHNi This doctoral thesis has been examined by a committee of the Division of Biological Engineering as follows: Associate Professor Bevin P. Engelward Chairman Professor Peter C. Dedon Supervisor Professor John M. Essigmann fl,(\ Dr. Richard J. Roberts --- Analysis of the Structural Changes Caused by Positive DNA Supercoiling by Marita Christine Barth Submitted to the Division of Biological Engineering on January 12, 2007 in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Macromolecular Biochemistry and Biophysics ABSTRACT The procession of helix-tracking enzymes along a DNA molecule results in the formation of supercoils in the DNA, with positive supercoiling (overwinding) generated ahead of the enzyme, and negative supercoiling (underwinding) in its wake. While the structural and physiological consequences of negative supercoiling have been well studied, technical challenges have prevented extensive examination of positively supercoiled DNA. Studies suggest that at sufficiently high levels of overwinding, DNA relieves strain by adopting an elongated structure, where the bases are positioned extrahelically and the backbones occupy the center of the helix. -

Assignment 1 Part I
1 Assignment 1 Note: Part I of the assignment is to be submitted through Blackboard (See instructions on Blackboard). Parts II, III and IV are to be submitted as a hard copy during your lab session. Part I Dilutions and Concentrations When doing your calculations, do not round off your intermediate numbers. Only round off the final answer. Your answers must be submitted to two significant figures after the decimal. For example 2.00, 0.020, or 0.0020. It is strongly recommended that you submit your answers using the web browser Firefox. You will be allowed two submissions! Use the following information to answer questions 1-6 You prepare a solution by adding the following ingredients in the order indicated: 600 mL H2O, 125 mL 1.6 M LiCl, 50 mL 20 % (m/v) MgCl2, and 25 mL 10g/L NaCl. The properties of each ingredient are as follows: LiCl: MW 200g/mole, density 1.2g/mL, density of 1.6 M soln. 1.05g/mL MgCl2: MW 150g/mole, density 1.3g/mL, density of 20% soln. 1.1g/mL NaCl: MW 35g/mole, density 1.15g/mL, density of 10g/L soln. 1.03g/mL Final solution: density: 1.25g/mL 1. What is the final molarity of MgCl2 in the solution? (0.083M) 2. What is the volume in milliliters of one part? (25 mL) 3. What is the percentage (m/m) of NaCl in the final solution? (0.025%) 4. What is the percentage (m/v) of LiCl in the final solution? (5%) 5. What is the number of parts of solvent in the final solution? (24 parts) 6. -

(Symposium-Workshop on Applications Of
OCCASION This publication has been made available to the public on the occasion of the 50th anniversary of the United Nations Industrial Development Organisation. DISCLAIMER This document has been produced without formal United Nations editing. The designations employed and the presentation of the material in this document do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations Industrial Development Organization (UNIDO) concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries, or its economic system or degree of development. Designations such as “developed”, “industrialized” and “developing” are intended for statistical convenience and do not necessarily express a judgment about the stage reached by a particular country or area in the development process. Mention of firm names or commercial products does not constitute an endorsement by UNIDO. FAIR USE POLICY Any part of this publication may be quoted and referenced for educational and research purposes without additional permission from UNIDO. However, those who make use of quoting and referencing this publication are requested to follow the Fair Use Policy of giving due credit to UNIDO. CONTACT Please contact [email protected] for further information concerning UNIDO publications. For more information about UNIDO, please visit us at www.unido.org UNITED NATIONS INDUSTRIAL DEVELOPMENT ORGANIZATION Vienna International Centre, P.O. Box 300, 1400 Vienna, Austria Tel: (+43-1) 26026-0 · www.unido.org · [email protected] 2f llf./ ?J~AL f.11e?OiPi~f of symposium-workshcp on Applications of Molecular Biological Research in Agriculture, Health and Environment 08-15 April, 1995, Lahore, Pakistan Prejeet N1.