Improving Metabarcoding Taxonomic Assignment: a Case Study Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Practical Handbook for Determining the Ages of Gulf of Mexico And
A Practical Handbook for Determining the Ages of Gulf of Mexico and Atlantic Coast Fishes THIRD EDITION GSMFC No. 300 NOVEMBER 2020 i Gulf States Marine Fisheries Commission Commissioners and Proxies ALABAMA Senator R.L. “Bret” Allain, II Chris Blankenship, Commissioner State Senator District 21 Alabama Department of Conservation Franklin, Louisiana and Natural Resources John Roussel Montgomery, Alabama Zachary, Louisiana Representative Chris Pringle Mobile, Alabama MISSISSIPPI Chris Nelson Joe Spraggins, Executive Director Bon Secour Fisheries, Inc. Mississippi Department of Marine Bon Secour, Alabama Resources Biloxi, Mississippi FLORIDA Read Hendon Eric Sutton, Executive Director USM/Gulf Coast Research Laboratory Florida Fish and Wildlife Ocean Springs, Mississippi Conservation Commission Tallahassee, Florida TEXAS Representative Jay Trumbull Carter Smith, Executive Director Tallahassee, Florida Texas Parks and Wildlife Department Austin, Texas LOUISIANA Doug Boyd Jack Montoucet, Secretary Boerne, Texas Louisiana Department of Wildlife and Fisheries Baton Rouge, Louisiana GSMFC Staff ASMFC Staff Mr. David M. Donaldson Mr. Bob Beal Executive Director Executive Director Mr. Steven J. VanderKooy Mr. Jeffrey Kipp IJF Program Coordinator Stock Assessment Scientist Ms. Debora McIntyre Dr. Kristen Anstead IJF Staff Assistant Fisheries Scientist ii A Practical Handbook for Determining the Ages of Gulf of Mexico and Atlantic Coast Fishes Third Edition Edited by Steve VanderKooy Jessica Carroll Scott Elzey Jessica Gilmore Jeffrey Kipp Gulf States Marine Fisheries Commission 2404 Government St Ocean Springs, MS 39564 and Atlantic States Marine Fisheries Commission 1050 N. Highland Street Suite 200 A-N Arlington, VA 22201 Publication Number 300 November 2020 A publication of the Gulf States Marine Fisheries Commission pursuant to National Oceanic and Atmospheric Administration Award Number NA15NMF4070076 and NA15NMF4720399. -

CHECKLIST and BIOGEOGRAPHY of FISHES from GUADALUPE ISLAND, WESTERN MEXICO Héctor Reyes-Bonilla, Arturo Ayala-Bocos, Luis E
ReyeS-BONIllA eT Al: CheCklIST AND BIOgeOgRAphy Of fISheS fROm gUADAlUpe ISlAND CalCOfI Rep., Vol. 51, 2010 CHECKLIST AND BIOGEOGRAPHY OF FISHES FROM GUADALUPE ISLAND, WESTERN MEXICO Héctor REyES-BONILLA, Arturo AyALA-BOCOS, LUIS E. Calderon-AGUILERA SAúL GONzáLEz-Romero, ISRAEL SáNCHEz-ALCántara Centro de Investigación Científica y de Educación Superior de Ensenada AND MARIANA Walther MENDOzA Carretera Tijuana - Ensenada # 3918, zona Playitas, C.P. 22860 Universidad Autónoma de Baja California Sur Ensenada, B.C., México Departamento de Biología Marina Tel: +52 646 1750500, ext. 25257; Fax: +52 646 Apartado postal 19-B, CP 23080 [email protected] La Paz, B.C.S., México. Tel: (612) 123-8800, ext. 4160; Fax: (612) 123-8819 NADIA C. Olivares-BAñUELOS [email protected] Reserva de la Biosfera Isla Guadalupe Comisión Nacional de áreas Naturales Protegidas yULIANA R. BEDOLLA-GUzMáN AND Avenida del Puerto 375, local 30 Arturo RAMíREz-VALDEz Fraccionamiento Playas de Ensenada, C.P. 22880 Universidad Autónoma de Baja California Ensenada, B.C., México Facultad de Ciencias Marinas, Instituto de Investigaciones Oceanológicas Universidad Autónoma de Baja California, Carr. Tijuana-Ensenada km. 107, Apartado postal 453, C.P. 22890 Ensenada, B.C., México ABSTRACT recognized the biological and ecological significance of Guadalupe Island, off Baja California, México, is Guadalupe Island, and declared it a Biosphere Reserve an important fishing area which also harbors high (SEMARNAT 2005). marine biodiversity. Based on field data, literature Guadalupe Island is isolated, far away from the main- reviews, and scientific collection records, we pres- land and has limited logistic facilities to conduct scien- ent a comprehensive checklist of the local fish fauna, tific studies. -

1 Exon Probe Sets and Bioinformatics Pipelines for All Levels of Fish Phylogenomics
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.18.949735; this version posted February 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Exon probe sets and bioinformatics pipelines for all levels of fish phylogenomics 2 3 Lily C. Hughes1,2,3,*, Guillermo Ortí1,3, Hadeel Saad1, Chenhong Li4, William T. White5, Carole 4 C. Baldwin3, Keith A. Crandall1,2, Dahiana Arcila3,6,7, and Ricardo Betancur-R.7 5 6 1 Department of Biological Sciences, George Washington University, Washington, D.C., U.S.A. 7 2 Computational Biology Institute, Milken Institute of Public Health, George Washington 8 University, Washington, D.C., U.S.A. 9 3 Department of Vertebrate Zoology, National Museum of Natural History, Smithsonian 10 Institution, Washington, D.C., U.S.A. 11 4 College of Fisheries and Life Sciences, Shanghai Ocean University, Shanghai, China 12 5 CSIRO Australian National Fish Collection, National Research Collections of Australia, 13 Hobart, TAS, Australia 14 6 Sam Noble Oklahoma Museum of Natural History, Norman, O.K., U.S.A. 15 7 Department of Biology, University of Oklahoma, Norman, O.K., U.S.A. 16 17 *Corresponding author: Lily C. Hughes, [email protected]. 18 Current address: Department of Organismal Biology and Anatomy, University of Chicago, 19 Chicago, IL. 20 21 Keywords: Actinopterygii, Protein coding, Systematics, Phylogenetics, Evolution, Target 22 capture 23 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.18.949735; this version posted February 19, 2020. -

Drum and Croaker (Family Sciaenidae) Diversity in North Carolina
Drum and Croaker (Family Sciaenidae) Diversity in North Carolina The waters along and off the coast are where you will find 18 of the 19 species within the Family Sciaenidae (Table 1) known from North Carolina. Until recently, the 19th species and the only truly freshwater species in this family, Freshwater Drum, was found approximately 420 miles WNW from Cape Hatteras in the French Broad River near Hot Springs. Table 1. Species of drums and croakers found in or along the coast of North Carolina. Scientific Name/ Scientific Name/ American Fisheries Society Accepted Common Name American Fisheries Society Accepted Common Name Aplodinotus grunniens – Freshwater Drum Menticirrhus saxatilis – Northern Kingfish Bairdiella chrysoura – Silver Perch Micropogonias undulatus – Atlantic Croaker Cynoscion nebulosus – Spotted Seatrout Pareques acuminatus – High-hat Cynoscion nothus – Silver Seatrout Pareques iwamotoi – Blackbar Drum Cynoscion regalis – Weakfish Pareques umbrosus – Cubbyu Equetus lanceolatus – Jackknife-fish Pogonias cromis – Black Drum Larimus fasciatus – Banded Drum Sciaenops ocellatus – Red Drum Leiostomus xanthurus – Spot Stellifer lanceolatus – Star Drum Menticirrhus americanus – Southern Kingfish Umbrina coroides – Sand Drum Menticirrhus littoralis – Gulf Kingfish With so many species historically so well-known to recreational and commercial fishermen, to lay people, and their availability in seafood markets, it is not surprising that these 19 species are known by many local and vernacular names. Skimming through the ETYFish Project -

Sharkcam Fishes
SharkCam Fishes A Guide to Nekton at Frying Pan Tower By Erin J. Burge, Christopher E. O’Brien, and jon-newbie 1 Table of Contents Identification Images Species Profiles Additional Info Index Trevor Mendelow, designer of SharkCam, on August 31, 2014, the day of the original SharkCam installation. SharkCam Fishes. A Guide to Nekton at Frying Pan Tower. 5th edition by Erin J. Burge, Christopher E. O’Brien, and jon-newbie is licensed under the Creative Commons Attribution-Noncommercial 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc/4.0/. For questions related to this guide or its usage contact Erin Burge. The suggested citation for this guide is: Burge EJ, CE O’Brien and jon-newbie. 2020. SharkCam Fishes. A Guide to Nekton at Frying Pan Tower. 5th edition. Los Angeles: Explore.org Ocean Frontiers. 201 pp. Available online http://explore.org/live-cams/player/shark-cam. Guide version 5.0. 24 February 2020. 2 Table of Contents Identification Images Species Profiles Additional Info Index TABLE OF CONTENTS SILVERY FISHES (23) ........................... 47 African Pompano ......................................... 48 FOREWORD AND INTRODUCTION .............. 6 Crevalle Jack ................................................. 49 IDENTIFICATION IMAGES ...................... 10 Permit .......................................................... 50 Sharks and Rays ........................................ 10 Almaco Jack ................................................. 51 Illustrations of SharkCam -

Of the FLORIDA STATE MUSEUM Biological Sciences
2% - p.*' + 0.:%: 4.' 1%* B -944 3 =5. M.: - . * 18 . .,:i -/- JL J-1.4:7 - of the FLORIDA STATE MUSEUM Biological Sciences Volume 24 1979 Number 1 THE ORIGIN AND SEASONALITY OF THE FISH FAUNA ON A NEW JETTY IN THE NORTHEASTERN GULF OF MEXICO ROBERT W. HASTINGS *S 0 4 - ' In/ g. .f, i»-ly -.Id UNIVERSITY OF FLORIDA - GAINESVILLE Numbers of the Bulletin of the Florida State Museum, Biological Sciences, are pub- lished at irregular intervals. Volumes contain about 300 pages and are not necessarily completed in any one calendar year. John William Hardy, Editor Rhoda J. Rybak, Managing Editor Consultants for this issue: Robert L. Shipp Donald P. deSylva Communications concerning purchase or exchange of the publications and all manuscripts should be addressed to: Managing Editor, Bulletin; Florida State Museum; University of Florida; Gainesville, Florida 32611. Copyright © 1979 by the Florida State Museum of the University of Florida. This public document was promulgated at an annual cost of $3,589.40, or $3.589 per copy. It makes available to libraries, scholars, and all interested persons the results of researches in the natural sciences, emphasizing the circum-Caribbean region. Publication date: November 12, 1979 Price, $3.60 THE ORIGIN AND SEASONALITY OF THE FISH FAUNA ON A NEW JETTY IN THE NORTHEASTERN GULF OF MEXICO ROBERT W. HASTINGS1 SYNOPSIS: The establishment of the fish fauna on a new jetty at East Pass at the mouth of Choctawhatchee Bay, Okaloosa County, Florida, was studied from June, 1968, to January, 1971. Important components of the jetty fauna during its initial stages of development were: (a) original residents that exhibit some attraction to reef habitats, including some sand-beach inhabitants, several pelagic species, and a few ubiquitous estuarine species; and (b) reef fishes originating from permanent populations on offshore reefs. -
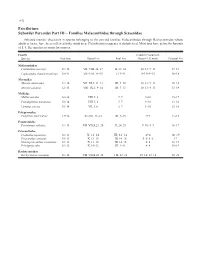
Perciformes Suborder Percoidei Part III – Families Malacanthidae
1152 Perciformes Suborder Percoidei Part III – Families Malacanthidae through Sciaenidae Selected meristic characters in species belonging to the percoid families Malacanthidae through Rachycentridae whose adults or larvae have been collected in the study area. Classification sequence is alphabetical. Most taxa have pelvic fin formula of I, 5. See species accounts for sources. Family Caudal (Procurrent, Species Vertebrae Dorsal Fin Anal Fin Dorsal + Ventral) Pectoral Fin Malacanthidae Caulolatilus microps 11+16 VII–VIII, 24–27 II, 22–24 10–13+9–13 17–18 Lopholatilus chamaeleonticeps 10+14 VII–VIII, 14–15 I, 13–14 9–13+9–13 16–18 Moronidae Morone americana 11+14 VII–XI, I, 11–13 III, 9–10 10–13+9–13 10–18 Morone saxatilis 12+13 VIII–IX, I, 9–14 III, 7–13 10–13+9–13 13–19 Mullidae Mullus auratus 10+14 VIII, I, 8 I, 7 9+10 15–17 Pseudupeneus maculatus 10+14 VIII, I, 8 I, 7 9+10 13–16 Upeneus parvus 10+14 VII, I, 8 I, 7 9+10 15–16 Polyprionidae Polyprion americanus 13+14 XI–XII, 11–12 III, 9–10 9+9 17–18 Pomatomidae Pomatomus saltatrix 11+15 VII–VIII,I,23–28 II, 24–29 9–10+8–9 16–17 Priacanthidae Cookeolus japonicus 10+13 X, 12–14 III, 12–14 4+4 18–19 Priacanthus arenatus 10+13 X, 13–15 III, 14–16 5–6+5–6 17 Heteropriacanthus cruentatus 10+13 X, 13–14 III, 14–15 4+4 18–19 Pristigenys alta 10+13 X, 10–12 III, 9–11 4+4 16–19 Rachycentridae Rachycentron canadum 11+14 VII–VIII,I,26–34 I–II, 22–28 15–16+12–14 20–21 Early Stages of Fishes in the Western North Atlantic Ocean 1153 Perciformes Suborder Percoidei Part III – Families Malacanthidae through Sciaenidae Selected meristic characters in species belonging to the percoid family Sciaenidae whose adults or larvae have been col- lected in the study area. -

Sciaenidae 1583
click for previous page Perciformes: Percoidei: Sciaenidae 1583 SCIAENIDAE Croakers (drums) by N.L. Chao, Universidade Federal do Amazonas, Manaus, Brazil iagnostic characters: Small to large (5 to 200 cm), most with fairly elongate and compressed body, few Dwith high body and fins (Equetus). Head short to medium-sized, usually with bony ridges on top of skull, cavernous canals visible externally in some (Stellifer, Nebris). Eye size variable, 1/9 to 1/3 in head length, some near-shore species with smaller eyes (Lonchurus, Nebris) and those mid- to deeper water ones with larger eyes (Ctenosciaena, Odontoscion).Mouth position and size extremely variable, from large, oblique with lower jaw projecting (Cynoscion) to small, inferior (Leiostomus) or with barbels (Paralonchurus). Sen- sory pores present at tip of snout (rostral pores, 3 to 7), and on lower margin of snout (marginal pores, 2 or 5). Tip of lower jaw (chin) with 2 to 6 mental pores, some with barbels, a single barbel (Menticirrhus), or in pairs along median edges of lower jaw (Micropogonias) or subopercles (Paralonchurus, Pogonias). Teeth usually small, villiform, set in bands on jaws with outer row of upper jaw and inner row of lower jaw slightly larger (Micropogonias), or on narrow bony ridges (Bairdiella); some with a pair of large ca- nines at the tip of upper jaw (Cynoscion, Isopisthus) or series of arrowhead canines on both jaws (Macrodon); roof of mouth toothless (no teeth on prevomer or palatine bones).Preopercle usually scaled, with or with- out spines or serration on -

61661147.Pdf
Resource Inventory of Marine and Estuarine Fishes of the West Coast and Alaska: A Checklist of North Pacific and Arctic Ocean Species from Baja California to the Alaska–Yukon Border OCS Study MMS 2005-030 and USGS/NBII 2005-001 Project Cooperation This research addressed an information need identified Milton S. Love by the USGS Western Fisheries Research Center and the Marine Science Institute University of California, Santa Barbara to the Department University of California of the Interior’s Minerals Management Service, Pacific Santa Barbara, CA 93106 OCS Region, Camarillo, California. The resource inventory [email protected] information was further supported by the USGS’s National www.id.ucsb.edu/lovelab Biological Information Infrastructure as part of its ongoing aquatic GAP project in Puget Sound, Washington. Catherine W. Mecklenburg T. Anthony Mecklenburg Report Availability Pt. Stephens Research Available for viewing and in PDF at: P. O. Box 210307 http://wfrc.usgs.gov Auke Bay, AK 99821 http://far.nbii.gov [email protected] http://www.id.ucsb.edu/lovelab Lyman K. Thorsteinson Printed copies available from: Western Fisheries Research Center Milton Love U. S. Geological Survey Marine Science Institute 6505 NE 65th St. University of California, Santa Barbara Seattle, WA 98115 Santa Barbara, CA 93106 [email protected] (805) 893-2935 June 2005 Lyman Thorsteinson Western Fisheries Research Center Much of the research was performed under a coopera- U. S. Geological Survey tive agreement between the USGS’s Western Fisheries -

Fishes of the Indian River Lagoon and Adjacent Waters, Florida
FISHES OF THE INDIAN RIVER LAGOON AND ADJACENT WATERS, FLORIDA by R. Grant Gilmore, Jr. Christopher J. Donohoe Douglas W. Cooke Harbor Branch Foundation, Inc. RR 1, Box 196 Fort Pierce, Florida 33450 and David J. Herrema Applied Biology, Inc. 641 DeKalb Industrial Way Decatur, Georgia 30033 Harbor Branch Foundation, Inc. Technical Report No. 41 September 1981 Funding was provided by the Harbor Branch Foundation, Inc. and Florida Power & Light Company, Miami, Florida FISHES OF THE INDIAN RIVER LAGOON AND ADJACENT WATERS, FLORIDA R. Grant Gilmore, Jr. Christopher Donohoe Dougl as Cooke Davi d Herrema INTRODUCTION It is the intent of this presentation to briefly describe regional fish habitats and to list the fishes associated with these habitats in the Indian River lagoon, its freshwater tributaries and the adjacent continental shelf to a depth of 200 m. A brief historical review of other regional ichthyological studies is also given. Data presented here revises the first regional description and checklist of fishes in east central Florida (Gilmore, 1977). The Indian River is a narrow estuarine lagoon system extending from Ponce de Leon Inlet in Vol usia County south to Jupiter Inlet in Palm Beach County (Fig. 1). It lies within the zone of overlap between two well known faunal regimes (i.e. the warm temperate Carolinian and the tropical Caribbean). To the north of the region, Hildebrand and Schroeder (1928), Fowler (1945), Struhsaker (1969), Dahlberg (1971), and others have made major icthyofaunal reviews of the coastal waters of the southeastern United States. McLane (1955) and Tagatz (1967) have made extensive surveys of the fishes of the St. -

<I>Pareques Umbrosus</I>
BULLETIN OF MARINE SCIENCE, 65(3): 825–838, 1999 LARVAL AND JUVENILE DEVELOPMENT OF THE CUBBYU PAREQUES UMBROSUS WITH NOTES ON THE HIGH HAT PAREQUES ACUMINATUS LARVAE G. Joan Holt and Cecilia M. Riley ABSTRACT Larval and juvenile development of Pareques umbrosus is described from laboratory spawned and reared specimens. Eggs of P. umbrosus ranged from 1.08 to 1.38 mm diam- eter with a single, pigmented oil globule 0.23 mm diameter. Yolk-sac larvae averaged 2.82 mm NL and had heavy golden-green stellate chromatophores covering the trunk and tail in fresh specimens. Preserved larvae had scattered melanophores on the trunk, tail and yolk-sac. Transition to exogenous feeding occurred at 2.59 mm NL and melanistic pigmentation included a distinct band around the tail anterior to the tip of the notochord, another vertical swath from the dorsal finfold through the pectorals to the pelvic area, and a lateral stripe that extended from the snout through the eye to the cleithrum. Two preopercle spines appeared just prior to flexion at 4.67 mm SL. A continuous horizontal stripe the length of the trunk and tail first developed at about 8.16 mm SL. Metamorpho- sis to the juvenile stage occurred between 10.00 and 15.00 mm SL. Juveniles averaging 30.5 mm SL had a characteristic 3-stripe color pattern, and a darkly pigmented first dorsal with a white trailing edge. The first dorsal was 1.5 times longer than the body depth. The pigmented pelvic fin had white leading and trailing edges. All but the center longitudinal body stripe were narrower than the pupil in juveniles with 4 to 6-stripes. -

El Niño Drives a Widespread Ulcerative Skin Disease Outbreak in Galapagos Marine Fishes
University of Rhode Island DigitalCommons@URI Fisheries, Animal and Veterinary Sciences Faculty Publications Fisheries, Animal and Veterinary Sciences 11-9-2018 El Niño Drives a Widespread Ulcerative Skin Disease Outbreak in Galapagos Marine Fishes Robert W. Lamb Franz Smith Anaide W. Aued Pelayo Salinas-de-León Jenifer Suarez See next page for additional authors Follow this and additional works at: https://digitalcommons.uri.edu/favs_facpubs Citation/Publisher Attribution Lamb, R. W., Smith, F., Aued, A. W., Salinas-de-Leon, P., Suarez, J., Gomez-Chiarri, M, Smolowitz, R., Giray, C., Witman, J. D. (2018). El Niño drives a widespread ulcerative skin disease outbreak in Galapagos marine fshes. Scientific Reports 8:16602. doi:10.1038/s41598-018-34929-z Available at: http://dx.doi.org/10.1038/s41598-018-34929-z This Article is brought to you for free and open access by the Fisheries, Animal and Veterinary Sciences at DigitalCommons@URI. It has been accepted for inclusion in Fisheries, Animal and Veterinary Sciences Faculty Publications by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. Authors Robert W. Lamb, Franz Smith, Anaide W. Aued, Pelayo Salinas-de-León, Jenifer Suarez, Marta Gomez- Chiarri, Roxanna Smolowitz, Cem Giray, and Jon D. Witman This article is available at DigitalCommons@URI: https://digitalcommons.uri.edu/favs_facpubs/28 www.nature.com/scientificreports OPEN El Niño drives a widespread ulcerative skin disease outbreak in Galapagos marine fshes Received: 17 May 2018 Robert W. Lamb 1, Franz Smith1, Anaide W. Aued2, Pelayo Salinas-de-León3,4, Accepted: 29 October 2018 Jenifer Suarez5, Marta Gomez-Chiarri6, Roxanna Smolowitz7, Cem Giray8 & Jon D.