1 Exon Probe Sets and Bioinformatics Pipelines for All Levels of Fish Phylogenomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

§4-71-6.5 LIST of CONDITIONALLY APPROVED ANIMALS November
§4-71-6.5 LIST OF CONDITIONALLY APPROVED ANIMALS November 28, 2006 SCIENTIFIC NAME COMMON NAME INVERTEBRATES PHYLUM Annelida CLASS Oligochaeta ORDER Plesiopora FAMILY Tubificidae Tubifex (all species in genus) worm, tubifex PHYLUM Arthropoda CLASS Crustacea ORDER Anostraca FAMILY Artemiidae Artemia (all species in genus) shrimp, brine ORDER Cladocera FAMILY Daphnidae Daphnia (all species in genus) flea, water ORDER Decapoda FAMILY Atelecyclidae Erimacrus isenbeckii crab, horsehair FAMILY Cancridae Cancer antennarius crab, California rock Cancer anthonyi crab, yellowstone Cancer borealis crab, Jonah Cancer magister crab, dungeness Cancer productus crab, rock (red) FAMILY Geryonidae Geryon affinis crab, golden FAMILY Lithodidae Paralithodes camtschatica crab, Alaskan king FAMILY Majidae Chionocetes bairdi crab, snow Chionocetes opilio crab, snow 1 CONDITIONAL ANIMAL LIST §4-71-6.5 SCIENTIFIC NAME COMMON NAME Chionocetes tanneri crab, snow FAMILY Nephropidae Homarus (all species in genus) lobster, true FAMILY Palaemonidae Macrobrachium lar shrimp, freshwater Macrobrachium rosenbergi prawn, giant long-legged FAMILY Palinuridae Jasus (all species in genus) crayfish, saltwater; lobster Panulirus argus lobster, Atlantic spiny Panulirus longipes femoristriga crayfish, saltwater Panulirus pencillatus lobster, spiny FAMILY Portunidae Callinectes sapidus crab, blue Scylla serrata crab, Samoan; serrate, swimming FAMILY Raninidae Ranina ranina crab, spanner; red frog, Hawaiian CLASS Insecta ORDER Coleoptera FAMILY Tenebrionidae Tenebrio molitor mealworm, -

New Insights on the Sister Lineage of Percomorph Fishes with an Anchored Hybrid Enrichment Dataset
Molecular Phylogenetics and Evolution 110 (2017) 27–38 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev New insights on the sister lineage of percomorph fishes with an anchored hybrid enrichment dataset ⇑ Alex Dornburg a, , Jeffrey P. Townsend b,c,d, Willa Brooks a, Elizabeth Spriggs b, Ron I. Eytan e, Jon A. Moore f,g, Peter C. Wainwright h, Alan Lemmon i, Emily Moriarty Lemmon j, Thomas J. Near b,k a North Carolina Museum of Natural Sciences, Raleigh, NC, USA b Department of Ecology & Evolutionary Biology and Peabody Museum of Natural History, Yale University, New Haven, CT 06520, USA c Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA d Department of Biostatistics, Yale University, New Haven, CT 06510, USA e Marine Biology Department, Texas A&M University at Galveston, Galveston, TX 77554, USA f Florida Atlantic University, Wilkes Honors College, Jupiter, FL 33458, USA g Florida Atlantic University, Harbor Branch Oceanographic Institution, Fort Pierce, FL 34946, USA h Department of Evolution & Ecology, University of California, Davis, CA 95616, USA i Department of Scientific Computing, Florida State University, 400 Dirac Science Library, Tallahassee, FL 32306, USA j Department of Biological Science, Florida State University, 319 Stadium Drive, Tallahassee, FL 32306, USA k Peabody Museum of Natural History, Yale University, New Haven, CT 06520, USA article info abstract Article history: Percomorph fishes represent over 17,100 species, including several model organisms and species of eco- Received 12 April 2016 nomic importance. Despite continuous advances in the resolution of the percomorph Tree of Life, resolu- Revised 22 February 2017 tion of the sister lineage to Percomorpha remains inconsistent but restricted to a small number of Accepted 25 February 2017 candidate lineages. -

Pacific Plate Biogeography, with Special Reference to Shorefishes
Pacific Plate Biogeography, with Special Reference to Shorefishes VICTOR G. SPRINGER m SMITHSONIAN CONTRIBUTIONS TO ZOOLOGY • NUMBER 367 SERIES PUBLICATIONS OF THE SMITHSONIAN INSTITUTION Emphasis upon publication as a means of "diffusing knowledge" was expressed by the first Secretary of the Smithsonian. In his formal plan for the Institution, Joseph Henry outlined a program that included the following statement: "It is proposed to publish a series of reports, giving an account of the new discoveries in science, and of the changes made from year to year in all branches of knowledge." This theme of basic research has been adhered to through the years by thousands of titles issued in series publications under the Smithsonian imprint, commencing with Smithsonian Contributions to Knowledge in 1848 and continuing with the following active series: Smithsonian Contributions to Anthropology Smithsonian Contributions to Astrophysics Smithsonian Contributions to Botany Smithsonian Contributions to the Earth Sciences Smithsonian Contributions to the Marine Sciences Smithsonian Contributions to Paleobiology Smithsonian Contributions to Zoo/ogy Smithsonian Studies in Air and Space Smithsonian Studies in History and Technology In these series, the Institution publishes small papers and full-scale monographs that report the research and collections of its various museums and bureaux or of professional colleagues in the world cf science and scholarship. The publications are distributed by mailing lists to libraries, universities, and similar institutions throughout the world. Papers or monographs submitted for series publication are received by the Smithsonian Institution Press, subject to its own review for format and style, only through departments of the various Smithsonian museums or bureaux, where the manuscripts are given substantive review. -

CHECKLIST and BIOGEOGRAPHY of FISHES from GUADALUPE ISLAND, WESTERN MEXICO Héctor Reyes-Bonilla, Arturo Ayala-Bocos, Luis E
ReyeS-BONIllA eT Al: CheCklIST AND BIOgeOgRAphy Of fISheS fROm gUADAlUpe ISlAND CalCOfI Rep., Vol. 51, 2010 CHECKLIST AND BIOGEOGRAPHY OF FISHES FROM GUADALUPE ISLAND, WESTERN MEXICO Héctor REyES-BONILLA, Arturo AyALA-BOCOS, LUIS E. Calderon-AGUILERA SAúL GONzáLEz-Romero, ISRAEL SáNCHEz-ALCántara Centro de Investigación Científica y de Educación Superior de Ensenada AND MARIANA Walther MENDOzA Carretera Tijuana - Ensenada # 3918, zona Playitas, C.P. 22860 Universidad Autónoma de Baja California Sur Ensenada, B.C., México Departamento de Biología Marina Tel: +52 646 1750500, ext. 25257; Fax: +52 646 Apartado postal 19-B, CP 23080 [email protected] La Paz, B.C.S., México. Tel: (612) 123-8800, ext. 4160; Fax: (612) 123-8819 NADIA C. Olivares-BAñUELOS [email protected] Reserva de la Biosfera Isla Guadalupe Comisión Nacional de áreas Naturales Protegidas yULIANA R. BEDOLLA-GUzMáN AND Avenida del Puerto 375, local 30 Arturo RAMíREz-VALDEz Fraccionamiento Playas de Ensenada, C.P. 22880 Universidad Autónoma de Baja California Ensenada, B.C., México Facultad de Ciencias Marinas, Instituto de Investigaciones Oceanológicas Universidad Autónoma de Baja California, Carr. Tijuana-Ensenada km. 107, Apartado postal 453, C.P. 22890 Ensenada, B.C., México ABSTRACT recognized the biological and ecological significance of Guadalupe Island, off Baja California, México, is Guadalupe Island, and declared it a Biosphere Reserve an important fishing area which also harbors high (SEMARNAT 2005). marine biodiversity. Based on field data, literature Guadalupe Island is isolated, far away from the main- reviews, and scientific collection records, we pres- land and has limited logistic facilities to conduct scien- ent a comprehensive checklist of the local fish fauna, tific studies. -

Subodha K. KARNA1, George N. KATSELIS2*, and Laith A. JAWAD3
ACTA ICHTHYOLOGICA ET PISCATORIA (2018) 48 (1): 83–86 DOI: 10.3750/AIEP/02259 LENGTH–WEIGHT RELATIONS OF 24 FISH SPECIES (ACTINOPTERYGII) FROM HIRAKUD RESERVOIR, ODISHA STATE OF INDIA Subodha K. KARNA1, George N. KATSELIS2*, and Laith A. JAWAD3 1 ICAR—Central Inland Fisheries Research Institute, Barrackpore, Kolkata, India 2 Department of Fisheries-Aquaculture Technology, Technological Educational Institute of Western Greece, 30200, Mesolonghi, Greece 34 Tinturn Place, Flat Bush, Manukau, Auckland 2016, New Zealand Karna S.K., Katselis G.N., Jawad L.A. 2018. Length–weight relations of 24 fish species (Actinopterygii) from Hirakud Reservoir, Odisha State of India. Acta Ichthyol. Piscat. 48 (1): 83–86. Abstract. Length–weight relations were estimated for 24 fish species sampled from the Hirakud Reservoir (Odisha State, India): Salmostoma bacaila (Hamilton, 1822); Salmostoma phulo (Hamilton, 1822); Labeo rohita (Hamilton, 1822); Labeo bata (Hamilton, 1822); Cirrhinus reba (Hamilton, 1822); Labeo calbasu (Hamilton, 1822); Puntius sophore (Hamilton, 1822); Puntius chola (Hamilton, 1822); Pethia ticto (Hamilton, 1822); Systomus sarana (Hamilton, 1822); Pethia phutunio (Hamilton, 1822); Osteobrama cotio (Hamilton, 1822); Amblypharyngodon mola (Hamilton, 1822); Rasbora rasbora (Hamilton, 1822); Parambassis ranga (Hamilton, 1822); Parambassis lala (Hamilton, 1822); Channa punctata (Bloch, 1793); Macrognathus pancalus (Hamilton, 1822); Notopterus notopterus (Pallas, 1769); Chanda nama (Hamilton, 1822); Xenentodon cancila (Hamilton, 1822); Glossogobius giuris (Hamilton, 1822); Ompok bimaculatus (Bloch, 1794); Gudusia chapra (Hamilton, 1822). They represented 10 families: Cyprinidae (14 species), Ambassidae (2 species), Channidae, Mastacembelidae, Notopteridae, Centropomidae, Belonidae, Gobiidae, Siluridae, and Clupeidae (1 species each). The b values ranged from 2.62 to 3.44. Nine of the species displayed isometric growth (b = 3), seven species negative allometric growth (b < 3), and eight species represented positive allometric growth (b < 3). -

Transcriptome Ortholog Alignment Sequence Tools (TOAST) for Phylogenomic Dataset Assembly
Transcriptome Ortholog Alignment Sequence Tools (TOAST) for Phylogenomic Dataset Assembly Dustin J. Wcisel North Carolina State University J. Thomas Howard North Carolina State University Jeffrey A. Yoder North Carolina State University Alex Dornburg ( [email protected] ) NC Museum of Natural Sciences https://orcid.org/0000-0003-0863-2283 Software Keywords: BUSCO ortholog assembly, Cetacean and teleost sh phylogeny, Missing Data Visualization, Transcriptome, Concatenated Alignment Posted Date: March 12th, 2020 DOI: https://doi.org/10.21203/rs.2.16269/v4 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Evolutionary Biology on March 30th, 2020. See the published version at https://doi.org/10.1186/s12862-020-01603-w. Page 1/18 Abstract Background Advances in next-generation sequencing technologies have reduced the cost of whole transcriptome analyses, allowing characterization of non-model species at unprecedented levels. The rapid pace of transcriptomic sequencing has driven the public accumulation of a wealth of data for phylogenomic analyses, however lack of tools aimed towards phylogeneticists to eciently identify orthologous sequences currently hinders effective harnessing of this resource. Results We introduce TOAST, an open source R software package that can utilize the ortholog searches based on the software Benchmarking Universal Single-Copy Orthologs (BUSCO) to assemble multiple sequence alignments of orthologous loci from transcriptomes for any group of organisms. By streamlining search, query, and alignment, TOAST automates the generation of locus and concatenated alignments, and also presents a series of outputs from which users can not only explore missing data patterns across their alignments, but also reassemble alignments based on user-dened acceptable missing data levels for a given research question. -

Table S1.Xlsx
Bone type Bone type Taxonomy Order/series Family Valid binomial Outdated binomial Notes Reference(s) (skeletal bone) (scales) Actinopterygii Incertae sedis Incertae sedis Incertae sedis †Birgeria stensioei cellular this study †Birgeria groenlandica cellular Ørvig, 1978 †Eurynotus crenatus cellular Goodrich, 1907; Schultze, 2016 †Mimipiscis toombsi †Mimia toombsi cellular Richter & Smith, 1995 †Moythomasia sp. cellular cellular Sire et al., 2009; Schultze, 2016 †Cheirolepidiformes †Cheirolepididae †Cheirolepis canadensis cellular cellular Goodrich, 1907; Sire et al., 2009; Zylberberg et al., 2016; Meunier et al. 2018a; this study Cladistia Polypteriformes Polypteridae †Bawitius sp. cellular Meunier et al., 2016 †Dajetella sudamericana cellular cellular Gayet & Meunier, 1992 Erpetoichthys calabaricus Calamoichthys sp. cellular Moss, 1961a; this study †Pollia suarezi cellular cellular Meunier & Gayet, 1996 Polypterus bichir cellular cellular Kölliker, 1859; Stéphan, 1900; Goodrich, 1907; Ørvig, 1978 Polypterus delhezi cellular this study Polypterus ornatipinnis cellular Totland et al., 2011 Polypterus senegalus cellular Sire et al., 2009 Polypterus sp. cellular Moss, 1961a †Scanilepis sp. cellular Sire et al., 2009 †Scanilepis dubia cellular cellular Ørvig, 1978 †Saurichthyiformes †Saurichthyidae †Saurichthys sp. cellular Scheyer et al., 2014 Chondrostei †Chondrosteiformes †Chondrosteidae †Chondrosteus acipenseroides cellular this study Acipenseriformes Acipenseridae Acipenser baerii cellular Leprévost et al., 2017 Acipenser gueldenstaedtii -

Monophyly and Interrelationships of Snook and Barramundi (Centropomidae Sensu Greenwood) and five New Markers for fish Phylogenetics ⇑ Chenhong Li A, , Betancur-R
Molecular Phylogenetics and Evolution 60 (2011) 463–471 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Monophyly and interrelationships of Snook and Barramundi (Centropomidae sensu Greenwood) and five new markers for fish phylogenetics ⇑ Chenhong Li a, , Betancur-R. Ricardo b, Wm. Leo Smith c, Guillermo Ortí b a School of Biological Sciences, University of Nebraska, Lincoln, NE 68588-0118, USA b Department of Biological Sciences, The George Washington University, Washington, DC 200052, USA c The Field Museum, Department of Zoology, Fishes, 1400 South Lake Shore Drive, Chicago, IL 60605, USA article info abstract Article history: Centropomidae as defined by Greenwood (1976) is composed of three genera: Centropomus, Lates, and Received 24 January 2011 Psammoperca. But composition and monophyly of this family have been challenged in subsequent Revised 3 May 2011 morphological studies. In some classifications, Ambassis, Siniperca and Glaucosoma were added to the Accepted 5 May 2011 Centropomidae. In other studies, Lates + Psammoperca were excluded, restricting the family to Available online 12 May 2011 Centropomus. Recent analyses of DNA sequences did not solve the controversy, mainly due to limited taxonomic or character sampling. The present study is based on DNA sequence data from thirteen Keywords: genes (one mitochondrial and twelve nuclear markers) for 57 taxa, representative of all relevant Centropomidae species. Five of the nuclear markers are new for fish phylogenetic studies. The monophyly of Centrop- Lates Psammoperca omidae sensu Greenwood was supported by both maximum likelihood and Bayesian analyses of a Ambassidae concatenated data set (12,888 bp aligned). No support was found for previous morphological hypothe- Niphon spinosus ses suggesting that ambassids are closely allied to the Centropomidae. -

Updated Checklist of Marine Fishes (Chordata: Craniata) from Portugal and the Proposed Extension of the Portuguese Continental Shelf
European Journal of Taxonomy 73: 1-73 ISSN 2118-9773 http://dx.doi.org/10.5852/ejt.2014.73 www.europeanjournaloftaxonomy.eu 2014 · Carneiro M. et al. This work is licensed under a Creative Commons Attribution 3.0 License. Monograph urn:lsid:zoobank.org:pub:9A5F217D-8E7B-448A-9CAB-2CCC9CC6F857 Updated checklist of marine fishes (Chordata: Craniata) from Portugal and the proposed extension of the Portuguese continental shelf Miguel CARNEIRO1,5, Rogélia MARTINS2,6, Monica LANDI*,3,7 & Filipe O. COSTA4,8 1,2 DIV-RP (Modelling and Management Fishery Resources Division), Instituto Português do Mar e da Atmosfera, Av. Brasilia 1449-006 Lisboa, Portugal. E-mail: [email protected], [email protected] 3,4 CBMA (Centre of Molecular and Environmental Biology), Department of Biology, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. E-mail: [email protected], [email protected] * corresponding author: [email protected] 5 urn:lsid:zoobank.org:author:90A98A50-327E-4648-9DCE-75709C7A2472 6 urn:lsid:zoobank.org:author:1EB6DE00-9E91-407C-B7C4-34F31F29FD88 7 urn:lsid:zoobank.org:author:6D3AC760-77F2-4CFA-B5C7-665CB07F4CEB 8 urn:lsid:zoobank.org:author:48E53CF3-71C8-403C-BECD-10B20B3C15B4 Abstract. The study of the Portuguese marine ichthyofauna has a long historical tradition, rooted back in the 18th Century. Here we present an annotated checklist of the marine fishes from Portuguese waters, including the area encompassed by the proposed extension of the Portuguese continental shelf and the Economic Exclusive Zone (EEZ). The list is based on historical literature records and taxon occurrence data obtained from natural history collections, together with new revisions and occurrences. -

Playfish Playfish
SpeSpecialeciale CiclidiCiclidi PPLAYLAYFFISHISH Collaborano alla diffusione di Playfi sh: AFAE - AIAM - AIG Copadichromis AIK - AIPC - AT - AVS borley “mdoka” CICHLIDPOWER CIR - DCI - GAB GAEM - GAF GAP - GARB GAS - OC Copadichromis sp. Yellow Jumbo Copadichromis trevavasae Nyassachromis prostoma Copadichromis ilesi nkanda irregolari irregolari sce ad intervalli ad sce Foto di Vlado Arnautovic Vlado di Foto Anno 2006 Anno Diffusione libera e gratuita e libera Diffusione proprio in Stampato E Playfi sh Speciale Ciclidi Cari amici di Playfi sh, ringraziarli a nome di tutti gli ac- Questo è il secondo numero spe- quariofi li che seguono l’iniziativa Speciale ciale per quest’anno, dedicato inte- (che sono davvero tanti). ramente alla revisione del genere Vi sorprenderebbe sapere il nu- Copadichromis. Il tutto è frutto del mero di copie scaricate media- lavoro di una squadra affi atata, mente per ogni numero. Il numero alla quale per l’occasione si sono speciale AIPC, poi, è fi nito dritto aggiunti Gianmarco e Ivan, pro- dritto su un forum tedesco, mentre venienti dal forum “Cichlidpower” alcuni forum centroamericani mi La redazione di Playfi sh (www.cichlidpower.it), che da oggi chiedono se è possibile la traduzio- impaginazione entra di diritto a far parte dei so- ne in lingua spagnola. Graziano Fiocca stenitori di Playfi sh. Tutto ciò fa enorme piacere, ma correzione bozze e grafi ca Ivan Salvatori è l’autore di que- preferisco, e con me lo Staff, “vo- Chiara di Biase sto corposo lavoro di traduzione lare basso”, perchè Playfi sh è nato copertina e ricerca, mentre Gianmarco si è come un gioco e tale deve restare; Carlo Carraro Graziano Fiocca “sbattuto” come pochi per riuscire certo, mira sempre a raggiungere ad ottenere tutti i permessi per l’uso il massimo della qualità possibile, revisione Graziella Antonello e la pubblicazione delle foto e delle ma senza dimenticare che siamo Enrico Carraro immagini. -

Automated Measurement of Long-Term Bower Behaviors in Lake Malawi
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.27.968511; this version posted February 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 1 Automated measurement of long-term bower behaviors in Lake Malawi cichlids using 2 depth sensing and action recognition 3 4 Zachary V Johnson1, Lijiang Long1,2, Junyu Li1, Manu Tej Sharma Arrojwala1, Vineeth Aljapur1, 5 Tyrone Lee1, Mark C Lowder1, Karen Gu1, Tucker J Lancaster1,2, Joseph I Stockert1, Jean M 6 Moorman3, Rachel L Lecesne4, Jeffrey T Streelman# 1,2,3, and Patrick T McGrath# 1,2,3,5 7 8 1School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA 30332, USA 9 2Interdisciplinary Graduate Program in Quantitative Biosciences, Georgia Institute of Technology, 10 Atlanta, GA 30332, USA 11 3Parker H. Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, 12 Atlanta, GA 30332, USA 13 4School of Computer Science, Georgia Institute of Technology, Atlanta, GA 30332, USA 14 5School of Physics, Georgia Institute of Technology, Atlanta, GA 30332, USA 15 16 #Co-corresponding authors: [email protected] (P.T.M.), 17 [email protected] (J.T.S.) 18 19 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.27.968511; this version posted February 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
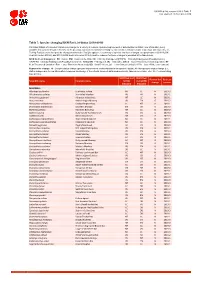
Table 7: Species Changing IUCN Red List Status (2018-2019)
IUCN Red List version 2019-3: Table 7 Last Updated: 10 December 2019 Table 7: Species changing IUCN Red List Status (2018-2019) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2018 (IUCN Red List version 2018-2) and 2019 (IUCN Red List version 2019-3) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered [CR(PE) - Critically Endangered (Possibly Extinct), CR(PEW) - Critically Endangered (Possibly Extinct in the Wild)], EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2018) List (2019) change version Category