Transcriptome Ortholog Alignment Sequence Tools (TOAST) for Phylogenomic Dataset Assembly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

§4-71-6.5 LIST of CONDITIONALLY APPROVED ANIMALS November
§4-71-6.5 LIST OF CONDITIONALLY APPROVED ANIMALS November 28, 2006 SCIENTIFIC NAME COMMON NAME INVERTEBRATES PHYLUM Annelida CLASS Oligochaeta ORDER Plesiopora FAMILY Tubificidae Tubifex (all species in genus) worm, tubifex PHYLUM Arthropoda CLASS Crustacea ORDER Anostraca FAMILY Artemiidae Artemia (all species in genus) shrimp, brine ORDER Cladocera FAMILY Daphnidae Daphnia (all species in genus) flea, water ORDER Decapoda FAMILY Atelecyclidae Erimacrus isenbeckii crab, horsehair FAMILY Cancridae Cancer antennarius crab, California rock Cancer anthonyi crab, yellowstone Cancer borealis crab, Jonah Cancer magister crab, dungeness Cancer productus crab, rock (red) FAMILY Geryonidae Geryon affinis crab, golden FAMILY Lithodidae Paralithodes camtschatica crab, Alaskan king FAMILY Majidae Chionocetes bairdi crab, snow Chionocetes opilio crab, snow 1 CONDITIONAL ANIMAL LIST §4-71-6.5 SCIENTIFIC NAME COMMON NAME Chionocetes tanneri crab, snow FAMILY Nephropidae Homarus (all species in genus) lobster, true FAMILY Palaemonidae Macrobrachium lar shrimp, freshwater Macrobrachium rosenbergi prawn, giant long-legged FAMILY Palinuridae Jasus (all species in genus) crayfish, saltwater; lobster Panulirus argus lobster, Atlantic spiny Panulirus longipes femoristriga crayfish, saltwater Panulirus pencillatus lobster, spiny FAMILY Portunidae Callinectes sapidus crab, blue Scylla serrata crab, Samoan; serrate, swimming FAMILY Raninidae Ranina ranina crab, spanner; red frog, Hawaiian CLASS Insecta ORDER Coleoptera FAMILY Tenebrionidae Tenebrio molitor mealworm, -

1 Exon Probe Sets and Bioinformatics Pipelines for All Levels of Fish Phylogenomics
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.18.949735; this version posted February 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Exon probe sets and bioinformatics pipelines for all levels of fish phylogenomics 2 3 Lily C. Hughes1,2,3,*, Guillermo Ortí1,3, Hadeel Saad1, Chenhong Li4, William T. White5, Carole 4 C. Baldwin3, Keith A. Crandall1,2, Dahiana Arcila3,6,7, and Ricardo Betancur-R.7 5 6 1 Department of Biological Sciences, George Washington University, Washington, D.C., U.S.A. 7 2 Computational Biology Institute, Milken Institute of Public Health, George Washington 8 University, Washington, D.C., U.S.A. 9 3 Department of Vertebrate Zoology, National Museum of Natural History, Smithsonian 10 Institution, Washington, D.C., U.S.A. 11 4 College of Fisheries and Life Sciences, Shanghai Ocean University, Shanghai, China 12 5 CSIRO Australian National Fish Collection, National Research Collections of Australia, 13 Hobart, TAS, Australia 14 6 Sam Noble Oklahoma Museum of Natural History, Norman, O.K., U.S.A. 15 7 Department of Biology, University of Oklahoma, Norman, O.K., U.S.A. 16 17 *Corresponding author: Lily C. Hughes, [email protected]. 18 Current address: Department of Organismal Biology and Anatomy, University of Chicago, 19 Chicago, IL. 20 21 Keywords: Actinopterygii, Protein coding, Systematics, Phylogenetics, Evolution, Target 22 capture 23 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.18.949735; this version posted February 19, 2020. -
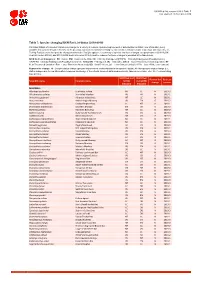
Table 7: Species Changing IUCN Red List Status (2018-2019)
IUCN Red List version 2019-3: Table 7 Last Updated: 10 December 2019 Table 7: Species changing IUCN Red List Status (2018-2019) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2018 (IUCN Red List version 2018-2) and 2019 (IUCN Red List version 2019-3) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered [CR(PE) - Critically Endangered (Possibly Extinct), CR(PEW) - Critically Endangered (Possibly Extinct in the Wild)], EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2018) List (2019) change version Category -

A Guide to the Parasites of African Freshwater Fishes
A Guide to the Parasites of African Freshwater Fishes Edited by T. Scholz, M.P.M. Vanhove, N. Smit, Z. Jayasundera & M. Gelnar Volume 18 (2018) Chapter 2.1. FISH DIVERSITY AND ECOLOGY Martin REICHARD Diversity of fshes in Africa Fishes are the most taxonomically diverse group of vertebrates and Africa shares a large portion of this diversity. This is due to its rich geological history – being a part of Gondwana, it shares taxa with the Neotropical region, whereas recent close geographical affnity to Eurasia permitted faunal exchange with European and Asian taxa. At the same time, relative isolation and the complex climatic and geological history of Africa enabled major diversifcation within the continent. The taxonomic diversity of African freshwater fshes is associated with functional and ecological diversity. While freshwater habitats form a tiny fraction of the total surface of aquatic habitats compared with the marine environment, most teleost fsh diversity occurs in fresh waters. There are over 3,200 freshwater fsh species in Africa and it is likely several hundreds of species remain undescribed (Snoeks et al. 2011). This high diversity and endemism is likely mirrored in diversity and endemism of their parasites. African fsh diversity includes an ancient group of air-breathing lungfshes (Protopterus spp.). Other taxa are capable of breathing air and tolerate poor water quality, including several clariid catfshes (e.g., Clarias spp.; Fig. 2.1.1D) and anabantids (Ctenopoma spp.). Africa is also home to several bichir species (Polypterus spp.; Fig. 2.1.1A), an ancient fsh group endemic to Africa, and bonytongue Heterotis niloticus (Cuvier, 1829) (Osteoglossidae), a basal actinopterygian fsh. -

A Review of the Systematic Biology of Fossil and Living Bony-Tongue Fishes, Osteoglossomorpha (Actinopterygii: Teleostei)
Neotropical Ichthyology, 16(3): e180031, 2018 Journal homepage: www.scielo.br/ni DOI: 10.1590/1982-0224-20180031 Published online: 11 October 2018 (ISSN 1982-0224) Copyright © 2018 Sociedade Brasileira de Ictiologia Printed: 30 September 2018 (ISSN 1679-6225) Review article A review of the systematic biology of fossil and living bony-tongue fishes, Osteoglossomorpha (Actinopterygii: Teleostei) Eric J. Hilton1 and Sébastien Lavoué2,3 The bony-tongue fishes, Osteoglossomorpha, have been the focus of a great deal of morphological, systematic, and evolutio- nary study, due in part to their basal position among extant teleostean fishes. This group includes the mooneyes (Hiodontidae), knifefishes (Notopteridae), the abu (Gymnarchidae), elephantfishes (Mormyridae), arawanas and pirarucu (Osteoglossidae), and the African butterfly fish (Pantodontidae). This morphologically heterogeneous group also has a long and diverse fossil record, including taxa from all continents and both freshwater and marine deposits. The phylogenetic relationships among most extant osteoglossomorph families are widely agreed upon. However, there is still much to discover about the systematic biology of these fishes, particularly with regard to the phylogenetic affinities of several fossil taxa, within Mormyridae, and the position of Pantodon. In this paper we review the state of knowledge for osteoglossomorph fishes. We first provide an overview of the diversity of Osteoglossomorpha, and then discuss studies of the phylogeny of Osteoglossomorpha from both morphological and molecular perspectives, as well as biogeographic analyses of the group. Finally, we offer our perspectives on future needs for research on the systematic biology of Osteoglossomorpha. Keywords: Biogeography, Osteoglossidae, Paleontology, Phylogeny, Taxonomy. Os peixes da Superordem Osteoglossomorpha têm sido foco de inúmeros estudos sobre a morfologia, sistemática e evo- lução, particularmente devido à sua posição basal dentre os peixes teleósteos. -

Systematic Morphology of Fishes in the Early 21St Century
Copeia 103, No. 4, 2015, 858–873 When Tradition Meets Technology: Systematic Morphology of Fishes in the Early 21st Century Eric J. Hilton1, Nalani K. Schnell2, and Peter Konstantinidis1 Many of the primary groups of fishes currently recognized have been established through an iterative process of anatomical study and comparison of fishes that has spanned a time period approaching 500 years. In this paper we give a brief history of the systematic morphology of fishes, focusing on some of the individuals and their works from which we derive our own inspiration. We further discuss what is possible at this point in history in the anatomical study of fishes and speculate on the future of morphology used in the systematics of fishes. Beyond the collection of facts about the anatomy of fishes, morphology remains extremely relevant in the age of molecular data for at least three broad reasons: 1) new techniques for the preparation of specimens allow new data sources to be broadly compared; 2) past morphological analyses, as well as new ideas about interrelationships of fishes (based on both morphological and molecular data) provide rich sources of hypotheses to test with new morphological investigations; and 3) the use of morphological data is not limited to understanding phylogeny and evolution of fishes, but rather is of broad utility to understanding the general biology (including phenotypic adaptation, evolution, ecology, and conservation biology) of fishes. Although in some ways morphology struggles to compete with the lure of molecular data for systematic research, we see the anatomical study of fishes entering into a new and exciting phase of its history because of recent technological and methodological innovations. -

Fish Diversity and Assemblages According to Distance from Source Along a Coastal River Gradient (Ehania River; South- East of Ivory Coast)
Iranian Journal of Fisheries Sciences 14(1)112-129 2015 Fish diversity and assemblages according to distance from source along a coastal river gradient (Ehania River; south- east of Ivory Coast) Konan K.F.1,2,3; Edia O.E.1; Bony K.Y.1,2; Kouamé K.M.1,3; Gourène G.1 Received: May 2013 Accepted: December 2014 Abstract Fish assemblage was investigated during the study of longitudinal profile of the Ehania River Basin in south-eastern Côte d’Ivoire. This area is subjected to intense human activities with many plantations (palm tree, banana, pineapple, coffee, rubber and cocoa). Samples were collected, with gillnets of different mesh sizes, through 6 sampling surveys during dry and rainy seasons from February 2010 to December 2010 at 6 sampling sites. A total of 70 fish species belonging to 48 genera, 28 families and 10 orders were recorded. The temporal variation of diversity index is less marked than spatial variation. The upstream, with 35 species, was less rich in species than the medium area and downstream areas (respectively 46 and 68). The upstream and downstream areas gathered 35 species. Thirty three species were common to the upper and middle areas and 46 species appeared both in the lower courses and the middle area. The 21 species restricted to the lower part of the river are mainly estuarine/marine origin. The beta diversity value revealed low similarity between the lower and upper course of Ehania River. The lowest Downloaded from jifro.ir at 13:43 +0330 on Tuesday October 5th 2021 values of Shannon’s diversity index and equitability index were observed in the middle part of the River which characterized by high population density and intense agricultural activity with many plantations. -

Life History Patterns and Biogeography: An
LIFE HISTORY PATTERNS AND Lynne R. Parenti2 BIOGEOGRAPHY: AN INTERPRETATION OF DIADROMY IN FISHES1 ABSTRACT Diadromy, broadly defined here as the regular movement between freshwater and marine habitats at some time during their lives, characterizes numerous fish and invertebrate taxa. Explanations for the evolution of diadromy have focused on ecological requirements of individual taxa, rarely reflecting a comparative, phylogenetic component. When incorporated into phylogenetic studies, center of origin hypotheses have been used to infer dispersal routes. The occurrence and distribution of diadromy throughout fish (aquatic non-tetrapod vertebrate) phylogeny are used here to interpret the evolution of this life history pattern and demonstrate the relationship between life history and ecology in cladistic biogeography. Cladistic biogeography has been mischaracterized as rejecting ecology. On the contrary, cladistic biogeography has been explicit in interpreting ecology or life history patterns within the broader framework of phylogenetic patterns. Today, in inferred ancient life history patterns, such as diadromy, we see remnants of previously broader distribution patterns, such as antitropicality or bipolarity, that spanned both marine and freshwater habitats. Biogeographic regions that span ocean basins and incorporate ocean margins better explain the relationship among diadromy, its evolution, and its distribution than do biogeographic regions centered on continents. Key words: Antitropical distributions, biogeography, diadromy, eels, -

18 Special Habitats and Special Adaptations 395
THE DIVERSITY OF FISHES Dedications: To our parents, for their encouragement of our nascent interest in things biological; To our wives – Judy, Sara, Janice, and RuthEllen – for their patience and understanding during the production of this volume; And to students and lovers of fishes for their efforts toward preserving biodiversity for future generations. Front cover photo: A Leafy Sea Dragon, Phycodurus eques, South Australia. Well camouflaged in their natural, heavily vegetated habitat, Leafy Sea Dragons are closely related to seahorses (Gasterosteiformes: Syngnathidae). “Leafies” are protected by Australian and international law because of their limited distribution, rarity, and popularity in the aquarium trade. Legal collection is highly regulated, limited to one “pregnant” male per year. See Chapters 15, 21, and 26. Photo by D. Hall, www.seaphotos.com. Back cover photos (from top to bottom): A school of Blackfin Barracuda, Sphyraena qenie (Perciformes, Sphyraenidae). Most of the 21 species of barracuda occur in schools, highlighting the observation that predatory as well as prey fishes form aggregations (Chapters 19, 20, 22). Blackfins grow to about 1 m length, display the silvery coloration typical of water column dwellers, and are frequently encountered by divers around Indo-Pacific reefs. Barracudas are fast-start predators (Chapter 8), and the pan-tropical Great Barracuda, Sphyraena barracuda, frequently causes ciguatera fish poisoning among humans (Chapter 25). Longhorn Cowfish, Lactoria cornuta (Tetraodontiformes: Ostraciidae), Papua New Guinea. Slow moving and seemingly awkwardly shaped, the pattern of flattened, curved, and angular trunk areas made possible by the rigid dermal covering provides remarkable lift and stability (Chapter 8). A Silvertip Shark, Carcharhinus albimarginatus (Carcharhiniformes: Carcharhinidae), with a Sharksucker (Echeneis naucrates, Perciformes: Echeneidae) attached. -

Adiaha Alda Alex UGWUMBA (Nee EKPO) (B) NATIONALITY: Nigerian
CURRICULUM VITAE 1. (a) NAME: Adiaha Alda Alex UGWUMBA (Nee EKPO) (b) NATIONALITY: Nigerian (c) STATE OF ORIGIN: Akwa Ibom State, Nigeria (d) DATE OF BIRTH: 15 September 1956 (e) MARITAL STATUS: Married (f) NO OF CHILDREN: Two (g) EMPLOYER: University of Ibadan DEPARTMENT: Zoology FACULTY: Science (h) PRESENT POST: Professor (2007) (i) POSTAL ADDRESS: Department of Zoology, University of Ibadan, Ibadan, Oyo State (j) HOME ADDRESS: 19 Amina Way, University of Ibadan, Ibadan, Nigeria (k) EMAIL ADDRESS: [email protected] [email protected] [email protected] (l) TELEPHONE NO.: +2348023416143; +2348097308117; (n) NEXT OF KIN AND ADDRESS: Prof. O. A. Ugwumba Department of Zoology University of Ibadan Ibadan, Nigeria III. UNIVERSITY EDUCATION: (a) University of Ibadan: Undergraduate, 1976-1979 Postgraduate (Ph.D), 1985-1990 (b) University of Lagos: Postgraduate (M.Sc), 1980-1982 IV. ACADEMIC QUALIFICATIONS (WITH DATES & GRANTING BODIES): (a) B. Sc. (Zoology), Second Class Upper Division, University of Ibadan July1979 (b) M. Sc. (Zoology) (Area of specialization: Fisheries), University of Lagos July 1983 (c) Ph. D. (Zoology) (Area of specialization: Fisheries), University of Ibadan July 1990 V. PROFESSIONAL QUALIFICATION AND DIPLOMA (WITH DATES): Federal Marine Fisheries School Certificate on Instruction for Fisheries/Research Officers Sept. 1983. VI. SCHOLARSHIPS, FELLOWSHIPS AND PRIZES IN THE LAST TEN YEARS: Nigeria University Doctoral Thesis Award Scheme (NUDAS) -Supervisor of Best Doctoral Thesis in Biological Sciences within the Nigeria University System in 2007 VII. HONOURS, DISTINCTIONS AND MEMBERSHIP OF LEARNED SOCIETIES: 1 a. Fellow, Fisheries Society of Nigeria (Ffs) b. Fellow, Zoological Society of Nigeria (Fzsn) c. Member, Nigerian Association of Aquatic Sciences d. -

Teleostei, Osteoglossiformes) in the Continental Lower Cretaceous of the Democratic Republic of Congo (Central Africa
Geo-Eco-Trop., 2015, 39, 2 : 247-254 On the presence of a second osteoglossid fish (Teleostei, Osteoglossiformes) in the continental Lower Cretaceous of the Democratic Republic of Congo (Central Africa) Sur la présence d’un second poisson ostéoglossidé (Teleostei, Osteoglossiformes) dans le Crétacé inférieur continental de la République Démocratique du Congo (Afrique centrale) Louis TAVERNE 1 Résumé: Un hyomandibulaire de téléostéen découvert dans les couches de la Formation de la Loia (Aptien- Albien continental) à Yakoko, sur la rivière Lomami, Province Centrale, République Démocratique du Congo, est décrit et ses relations phylogénétiques sont discutées. L’os est grand et porte un processus operculaire très allongé. Des comparaisons avec d’autres téléostéens du Crétacé inférieur continental indiquent que cet hyomandibulaire appartient à un ostéoglossidé qui semble proche de Paralycoptera. Mots-clés: Teleostei, Osteoglossidae, hyomandibulaire, Formation de la Loia, Crétacé inférieur continental, Yakoko, République Démocratique du Congo Abstract: A teleost hyomandibula discovered in the deposits of the Loia Formation (continental Aptian- Albian) at Yakoko, on the Lomami River, Central Province, Democratic Republic of Congo, is described and its phylogenetic relationships are discussed. The bone is rather large and bears an extremely long opercular process. Comparisons with other freshwater Early Cretaceous teleosts indicate that this hyomandibula belongs to an osteoglossid fish that seems close to Paralycoptera. Key words: Teleostei, Osteoglossidae, hyomandibula, Loia Formation, continental Early Cretaceous, Yakoko, Democratic Republic of Congo. INTRODUCTION The Loia and the Bokungu Formations are respectively the lower and the upper strata within the continental Early Cretaceous deposits of the Congolese Cuvette and the surrounding zones, in the Democratic Republic of Congo (CAHEN et al., 1959, 1960; CASIER, 1961). -

A Review of the Systematic Biology of Fossil and Living Bony-Tongue Fishes, Osteoglossomorpha (Actinopterygii: Teleostei)" (2018)
W&M ScholarWorks VIMS Articles Virginia Institute of Marine Science 2018 A review of the systematic biology of fossil and living bony- tongue fishes, Osteoglossomorpha (Actinopterygii: Teleostei) Eric J. Hilton Virginia Institute of Marine Science Sebastien Lavoue Follow this and additional works at: https://scholarworks.wm.edu/vimsarticles Part of the Aquaculture and Fisheries Commons Recommended Citation Hilton, Eric J. and Lavoue, Sebastien, "A review of the systematic biology of fossil and living bony-tongue fishes, Osteoglossomorpha (Actinopterygii: Teleostei)" (2018). VIMS Articles. 1297. https://scholarworks.wm.edu/vimsarticles/1297 This Article is brought to you for free and open access by the Virginia Institute of Marine Science at W&M ScholarWorks. It has been accepted for inclusion in VIMS Articles by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Neotropical Ichthyology, 16(3): e180031, 2018 Journal homepage: www.scielo.br/ni DOI: 10.1590/1982-0224-20180031 Published online: 11 October 2018 (ISSN 1982-0224) Copyright © 2018 Sociedade Brasileira de Ictiologia Printed: 30 September 2018 (ISSN 1679-6225) Review article A review of the systematic biology of fossil and living bony-tongue fishes, Osteoglossomorpha (Actinopterygii: Teleostei) Eric J. Hilton1 and Sébastien Lavoué2,3 The bony-tongue fishes, Osteoglossomorpha, have been the focus of a great deal of morphological, systematic, and evolutio- nary study, due in part to their basal position among extant teleostean fishes. This group includes the mooneyes (Hiodontidae), knifefishes (Notopteridae), the abu (Gymnarchidae), elephantfishes (Mormyridae), arawanas and pirarucu (Osteoglossidae), and the African butterfly fish (Pantodontidae). This morphologically heterogeneous group also has a long and diverse fossil record, including taxa from all continents and both freshwater and marine deposits.