TSC1/TSC2 Signaling in the CNS ⇑ Juliette M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

Lymphocyte/WBC-Based Comprehensive Testing Via Next-Gen Sequencing NF1/SPRED1 and Other Rasopathy Related Conditions on Blood/Saliva
Last Updated October 2019 Institutional Price TAT Genetic Test CPT Codes Z codes Specimen Requirements (USD$) (working days) Lymphocyte/WBC-based Comprehensive Testing via Next-Gen Sequencing NF1/SPRED1 and Other RASopathy Related Conditions on Blood/Saliva NF1- only NGS testing and copy number analysis for the NF1 gene (NF1-NG) This testing includes an average coverage of >1600x to allow for the identification of (1) 3-6ml whole blood in EDTA (purple topped) tubes mosaicism as low as 3-5% of the alleles. In addition, novel variants identified in the $1,000 81408 25 (2) Oragene 575 saliva kit (provided by the MGL) ZB6A9 NF1 gene will be confirmed via RNA-based analysis at no additional charge. RNA- $1,600 (RUSH) 81479 15 (RUSH) (3) DNA sample (25ul volume at 3ug, O.D. value at based testing will also be provided to non-founder, multigenerational families with 260:280 ≥1.8) “classic” NF1 at no additional charge if next-generation sequencing is found negative. (1) 3-6ml whole blood in EDTA (purple topped) tubes SPRED1-only NGS testing and copy number analysis for SPRED1 (SPD1-NG) $800 81405 25 (2) Oragene 575 saliva kit (provided by the MGL) This testing includes an average coverage of >1600x to allow for the identification of ZB6AC $1,400 (RUSH) 81479 15 (RUSH) (3) DNA sample (25ul volume at 3ug, O.D. value at mosaicism as low as 3-5% of the alleles after comprehensive NF1 analysis. 260:280 ≥1.8) NF1/SPRED1 NGS testing and copy number analysis for NF1 and SPRED1 (NFSP-NG) (1) 3-6ml whole blood in EDTA (purple topped) tubes 81408 This testing includes an average coverage of >1600x to allow for the identification of $1,100 25 (2) Oragene 575 saliva kit (provided by the MGL) 81405 ZB6A8 mosaicism as low as 3-5% of the alleles. -

Mtor: a Pharmacologic Target for Autophagy Regulation
mTOR: a pharmacologic target for autophagy regulation Young Chul Kim, Kun-Liang Guan J Clin Invest. 2015;125(1):25-32. https://doi.org/10.1172/JCI73939. Review mTOR, a serine/threonine kinase, is a master regulator of cellular metabolism. mTOR regulates cell growth and proliferation in response to a wide range of cues, and its signaling pathway is deregulated in many human diseases. mTOR also plays a crucial role in regulating autophagy. This Review provides an overview of the mTOR signaling pathway, the mechanisms of mTOR in autophagy regulation, and the clinical implications of mTOR inhibitors in disease treatment. Find the latest version: https://jci.me/73939/pdf The Journal of Clinical Investigation REVIEW SERIES: AUTOPHAGY Series Editor: Guido Kroemer mTOR: a pharmacologic target for autophagy regulation Young Chul Kim and Kun-Liang Guan Department of Pharmacology and Moores Cancer Center, UCSD, La Jolla, California, USA. mTOR, a serine/threonine kinase, is a master regulator of cellular metabolism. mTOR regulates cell growth and proliferation in response to a wide range of cues, and its signaling pathway is deregulated in many human diseases. mTOR also plays a crucial role in regulating autophagy. This Review provides an overview of the mTOR signaling pathway, the mechanisms of mTOR in autophagy regulation, and the clinical implications of mTOR inhibitors in disease treatment. Overview of mTOR signaling pathway such as insulin and IGF activate their cognate receptors (recep- Nutrients, growth factors, and cellular energy levels are key deter- tor tyrosine kinases [RTKs]) and subsequently activate the PI3K/ minants of cell growth and proliferation. mTOR, a serine/threon- AKT signaling axis. -

Mutational Spectrum of the TSC1 Gene in a Cohort of 225 Tuberous Sclerosis Complex Patients: J Med Genet: First Published As 10.1136/Jmg.36.4.285 on 1 April 1999
J Med Genet 1999;36:285–289 285 Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: J Med Genet: first published as 10.1136/jmg.36.4.285 on 1 April 1999. Downloaded from no evidence for genotype-phenotype correlation Marjon van Slegtenhorst, Senno Verhoef, Anita Tempelaars, Lida Bakker, Qi Wang, Marja Wessels, Remco Bakker, Mark Nellist, Dick Lindhout, Dicky Halley, Ans van den Ouweland Abstract TSC1 locus on chromosome 9q34 and the Tuberous sclerosis complex is an inherited other half to the TSC2 locus on chromosome tumour suppressor syndrome, caused by a 16p13.67 The TSC1 and TSC2 genes were mutation in either the TSC1 or TSC2 identified by positional cloning89 and there is gene. The disease is characterised by a abundant evidence that both genes act as broad phenotypic spectrum that can in- tumour suppressor genes.10–13 clude seizures, mental retardation, renal The TSC2 gene consists of 41 exons, dysfunction, and dermatological abnor- spanning 43 kb of genomic DNA.14 It encodes malities. The TSC1 gene was recently a 200 kDa protein, tuberin, which has a identified and has 23 exons, spanning 45 putative GAP activity for rab515 and rap1,16 two kb of genomic DNA, and encoding an 8.6 members of the ras superfamily of small kb mRNA. After screening all 21 coding GTPases. The mutational spectrum of TSC2 exons in our collection of 225 unrelated includes a number of large deletions often dis- 17 18 patients, only 29 small mutations were rupting the PKD1 gene as well, but also 19–27 detected, suggesting that TSC1 mutations point mutations and a number of missense 28 are under-represented among TSC pa- changes. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Immersion Course Lecture Kelly
Genomic Counseling in the research setting Kelly East, MS CGC Genetic Counselor HudsonAlpha Institute for Biotechnology single gene disorders • mutation in a single gene leads to disease • often has characteristic family inheritance patterns J Neurol Neurosurg Psychiatry 2002;73:ii5-ii11 Genes that code for hemoglobin 1 2 3 4 5 HBB 6 7 8 9 10 11 12 HBA2 13 14 15 16 17 18 19 20 21 22 X Y http://www.genome.gov/glossary/resources/karyotype.pdf TARDBP Genes associated ALS2 with ALS 1 2 3 4 5 C9ORF72 OPTN FIG4 ATXN2 SETX 6 7 8 9 10 11 12 ANG SPG11 FUS 13 14 15 16 17 18 VAPB UBQLN2 SOD1 19 20 21 22 X Y http://www.genome.gov/glossary/resources/karyotype.pdf complex traits • most common traits and diseases have a complex etiology • causative risks can include genetic changes (both large and small scale) environmental factors (head injury, nutrition, exposure to toxins) societal factors (death of family member, abuse, hardships) • in most cases - not triggered by a change in a single gene but rather by the interaction of several genetic, environmental and societal risks spectrum of human genetic conditions CGTATACCGGGTCATGCACGTGTAGAGCGAGTTAGCTCGCTGGCTAAAGAGGGTCGAC ATCCGCGAGTTTATGAGGAAGAATCGGCAGCTTGACCGAAGAGGCGTGGTAAGACCCG TTAGGGATCGTATACCGGGTCATGCACGTGTAGAGCGAGTTAGCTCGCTGGCTAAAGA GGGTCGACATCCGCGAGTTTATGAGGAAGAATCGGCAGCTTGACCGAAGAGGCGTGGT AAGACCCGTTAGGGATCGTATACCGGGTCATGCACGTGTAGAGCGAGTTAGCTCGCTG GCTAAAGAGGGTCGACATCCGCGAGTTTATGAGGAAGAATCGGCAGCTTGACCGAAGA GGCGTGGTAAGACCCGTTAGGGATCGTATACCGGGTCATGCACGTGTAGAGCGAGTTA in patients with a suspected genetic -

Tuberous Sclerosis Complex: Molecular Pathogenesis and Animal Models
Neurosurg Focus 20 (1):E4, 2006 Tuberous sclerosis complex: molecular pathogenesis and animal models LEANDRO R. PIEDIMONTE, M.D., IAN K. WAILES, M.D., AND HOWARD L. WEINER, M.D. Division of Pediatric Neurosurgery, Department of Neurosurgery, New York University School of Medicine, New York, New York Mutations in one of two genes, TSC1 and TSC2, result in a similar disease phenotype by disrupting the normal inter- action of their protein products, hamartin and tuberin, which form a functional signaling complex. Disruption of these genes in the brain results in abnormal cellular differentiation, migration, and proliferation, giving rise to the charac- teristic brain lesions of tuberous sclerosis complex (TSC) called cortical tubers. The most devastating complications of TSC affect the central nervous system and include epilepsy, mental retardation, autism, and glial tumors. Relevant animal models, including conventional and conditional knockout mice, are valuable tools for studying the normal func- tions of tuberin and hamartin and the way in which disruption of their expression gives rise to the variety of clinical features that characterize TSC. In the future, these animals will be invaluable preclinical models for the development of highly specific and efficacious treatments for children affected with TSC. KEY WORDS • tuberous sclerosis • pathogenesis • animal model Tuberous sclerosis complex is an autosomal-dominant zures, mental retardation, and autism.1,51 The number and tumor predisposition syndrome that affects approximately location of cortical tubers and the patient’s age at seizure 1 in 7500 individuals worldwide.19 The TSC is character- onset are highly correlated with neurological outcomes.1,30 ized by benign hamartomatous growths in multiple organs, Epilepsy occurs in approximately 80% of affected indi- including the kidney, skin, retina, lung, and brain. -

Rho Guanine Nucleotide Exchange Factors: Regulators of Rho Gtpase Activity in Development and Disease
Oncogene (2014) 33, 4021–4035 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease DR Cook1, KL Rossman2,3 and CJ Der1,2,3 The aberrant activity of Ras homologous (Rho) family small GTPases (20 human members) has been implicated in cancer and other human diseases. However, in contrast to the direct mutational activation of Ras found in cancer and developmental disorders, Rho GTPases are activated most commonly in disease by indirect mechanisms. One prevalent mechanism involves aberrant Rho activation via the deregulated expression and/or activity of Rho family guanine nucleotide exchange factors (RhoGEFs). RhoGEFs promote formation of the active GTP-bound state of Rho GTPases. The largest family of RhoGEFs is comprised of the Dbl family RhoGEFs with 70 human members. The multitude of RhoGEFs that activate a single Rho GTPase reflects the very specific role of each RhoGEF in controlling distinct signaling mechanisms involved in Rho activation. In this review, we summarize the role of Dbl RhoGEFs in development and disease, with a focus on Ect2 (epithelial cell transforming squence 2), Tiam1 (T-cell lymphoma invasion and metastasis 1), Vav and P-Rex1/2 (PtdIns(3,4,5)P3 (phosphatidylinositol (3,4,5)-triphosphate)-dependent Rac exchanger). Oncogene (2014) 33, 4021–4035; doi:10.1038/onc.2013.362; published online 16 September 2013 Keywords: Rac1; RhoA; Cdc42; guanine nucleotide exchange factors; cancer; -

Mir-126-3P Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Via Downregulating SPRED1
38 Original Article Page 1 of 14 miR-126-3p contributes to sorafenib resistance in hepatocellular carcinoma via downregulating SPRED1 Wenliang Tan1,2#, Zhirong Lin1,2#, Xianqing Chen3, Wenxin Li4, Sicong Zhu1,5, Yingcheng Wei1,2, Liyun Huo1,2, Yajin Chen1,2, Changzhen Shang1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 2Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 3Department of Hepatobiliary Surgery, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 4Department of Cardiology, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 5Department of Surgical Intensive Care Unit, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China Contributions: (I) Conception and design: C Shang, Y Chen; (II) Administrative support: C Shang, Y Chen; (III) Provision of study materials or patients: W Tan, Z Lin; (IV) Collection and assembly of data: S Zhu, Y Wei, L Huo; (V) Data analysis and interpretation: X Chen, W Li; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Changzhen Shang; Yajin Chen. Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou 510120, China. Email: [email protected]; [email protected]. Background: Sorafenib can prolong the survival of patients with advanced hepatocellular carcinoma (HCC). However, drug resistance remains the main obstacle to improving its efficiency. This study aimed to explore the likely molecular mechanism of sorafenib resistance. Methods: Differentially expressed microRNAs (miRNAs) related to sorafenib response were analyzed with the Limma package in R software. -

Neurofibromatosis-1 Regulates Mtor-Mediated Astrocyte Growth
Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner Sutapa Banerjee, Nikkilina R. Crouse, Ryan J. Emnett, Scott M. Gianino, and David H. Gutmann1 Department of Neurology, Washington University School of Medicine, St. Louis, MO 63110 Edited* by Webster K. Cavenee, Ludwig Institute, University of California at San Diego, La Jolla, CA, and approved August 15, 2011 (received for review December 17, 2010) Converging evidence from the analysis of human brain tumors seizures and death, whereas Nf1 loss in these same neuroglial and genetically engineered mice has revealed that the mamma- progenitors has no effect on mouse viability or epileptogenesis (16, lian target of rapamycin (mTOR) pathway is a central regulator of 17). Importantly, although Pten-, hamartin-, and neurofibromin- glial and glioma cell growth. In this regard, mutational inactiva- deficient glial phenotypes have different biological effects, each tion of neurofibromatosis-1 (NF1), tuberous sclerosis complex is completely inhibited by rapamycin in vitro and in vivo (15, 18– (TSC), and PTEN genes is associated with glioma formation, such 21). Collectively, these observations support a model in which fi that pharmacologic inhibition of mTOR signaling results in atten- neuro bromin-regulated mTOR-dependent growth regulation uated tumor growth. This shared dependence on mTOR suggests is distinct from TSC/Rheb-mediated mTOR-dependent growth that PTEN and NF1 (neurofibromin) glial growth regulation regulation in the brain. To -

Snapshot: Mtorc1 Signaling at the Lysosomal Surface Liron Bar-Peled and David M
1390 Cell SnapShot: mTORC1 Signaling at the 151 , December 7,2012©2012 ElsevierInc. DOI http://dx.doi.org/10.1016/j.cell.2012.11.038 Lysosomal Surface Liron Bar-Peled and David M. Sabatini Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA Nutrient signaling Wnt Growth Leu signaling factor signaling TNF signaling Amino Gln Wnt Tyrosine acids Frizzled IGF kinase TNFD receptor TNF receptor DNA Energy levels O levels SLC1A5 SLC7A5 2 damage ATP/AMP PIP2 Pten Amino acid mTORC1 Dsh1 IRS1 transporter Gln (inactive) GRB2 SOS PI3K PIP3 Gln Redd1 p53 LKB1 GEF GSK3 activity Leu GTP Ras NF1 PDK1 GAP Sestrin Movement to the activity RagAGTP lysosomal surface Movement AMPK Raf GDP away from Rapamycin RagA the lysosomal surface FKBP12 Mek Erk1/2 Rsk1 Akt1 IKK` v-ATPase GEF activity CYTOPLASM RagAGTP mTORC1 Ragulator GTP GAP activity GDP TSC complex RagC (active) Rheb Tumor suppressor Oncogene mTORC1 substrate ? Growth DOWNSTREAM CELLULAR PROGRAMS REGULATED BY mTORC1 ACTIVITY See online version for legend and references. S6K1 Amino acids LYSOSOME Protein synthesis 4EBP1 COMPLEXES AT THE LYSOSOMAL SURFACE HIF1_ Energy metabolism A B A E E G G pras40 deptor Lysosome biogenesis A BB MP1 HBXIP TSC1 TBC1D7 TFEB p14 C7orf59 raptor H mLST8 Lipin-1 D C Lipid p18 biosynthesis a d F mTOR TSC2 SREBP1/2 c cc ATG13 FIP200 Autophagy Lysosomal v-ATPase Ragulator complex mTORC1 TSC complex ULK1 SnapShot: mTORC1 Signaling at the Lysosomal Surface Liron Bar-Peled and David M. Sabatini Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA In mammals, the mTOR complex 1 (mTORC1) ser/thr kinase regulates cellular and organismal growth in response to a variety of environmental and intracellular stimuli. -
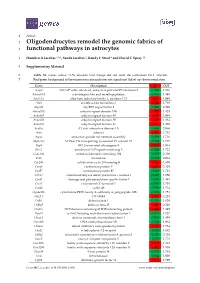
Oligodendrocytes Remodel the Genomic Fabrics of Functional Pathways in Astrocytes
1 Article 2 Oligodendrocytes remodel the genomic fabrics of 3 functional pathways in astrocytes 4 Dumitru A Iacobas 1,2,*, Sanda Iacobas 3, Randy F Stout 4 and David C Spray 2,5 5 Supplementary Material 6 Table S1. Genes whose >1.5x absolute fold-change did not meet the individual CUT criterion. 7 Red/green background of the expression ratio indicates not significant (false) up-/down-regulation. Gene Description X CUT Acap2 ArfGAP with coiled-coil, ankyrin repeat and PH domains 2 -1.540 1.816 Adamts18 a disintegrin-like and metallopeptidase -1.514 1.594 Akr1c12 aldo-keto reductase family 1, member C12 1.866 1.994 Alx3 aristaless-like homeobox 3 1.536 1.769 Alyref2 Aly/REF export factor 2 -1.880 2.208 Ankrd33b ankyrin repeat domain 33B 1.593 1.829 Ankrd45 ankyrin repeat domain 45 1.514 1.984 Ankrd50 ankyrin repeat domain 50 1.628 1.832 Ankrd61 ankyrin repeat domain 61 1.645 1.802 Arid1a AT rich interactive domain 1A -1.668 2.066 Artn artemin 1.524 1.732 Aspm abnormal spindle microtubule assembly -1.693 1.716 Atp6v1e1 ATPase, H+ transporting, lysosomal V1 subunit E1 -1.679 1.777 Bag4 BCL2-associated athanogene 4 1.723 1.914 Birc3 baculoviral IAP repeat-containing 3 -1.588 1.722 Ccdc104 coiled-coil domain containing 104 -1.819 2.130 Ccl2 chemokine -1.699 2.034 Cdc20b cell division cycle 20 homolog B 1.512 1.605 Cenpf centromere protein F 2.041 2.128 Cep97 centrosomal protein 97 -1.641 1.723 COX1 mitochondrially encoded cytochrome c oxidase I -1.607 1.650 Cpsf7 cleavage and polyadenylation specific factor 7 -1.635 1.891 Crct1 cysteine-rich