Topics in Phosphorus-Flij Orine Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Ereztech LLC P3553 Safety Data Sheet
EREZTECH LLC 11555 Medlock Bridge Road, Suite 100, Johns Creek, GA 30097, USA T: +1.888.658.1221 F: 1.678.619.2020 E: [email protected] W: https://ereztech.com Section 1. Identification Product Name: Phosphorus trifluoride Product Type: Gas CAS Number: 7783-55-3 Product Number: P3553 Product Manufacturer: Ereztech LLC 11555 Medlock Bridge Road, Suite 100 Johns Creek, GA 30097 Product Information: (888) 658-1221 In Case of an Emergency: CHEMTREC: 1-800-424-9300 (USA); +1 703-527-3887 (International); CCN836180 *** Contact manufacturer for all non-emergency calls. Section 2. Hazards Identification Appearance/Odor: Colorless, odorless gas. Classification: ACUTE TOXICITY, ORAL - Category 4, H302 ACUTE TOXICITY, DERMAL - Category 4, H312 SKIN CORROSION/IRRITATION - Category 1B, H314 SERIOUS EYE DAMAGE/EYE IRRITATION - Category 2, H318 ACUTE TOXICITY, INHALATION - Category 1, H330 SPECIFIC TARGET ORGAN TOXICITY, SINGLE EXPOSURE; RESPIRATORY TRACT IRRITATION – Category 3, H335 GHS Label Elements Hazard Pictograms: Signal Word: DANGER Hazard Statements: H302: Harmful if swallowed. H312: Harmful in contact with skin. H314: Causes severe skin burns and eye damage. H318: Causes serious eye damage. H330: Fatal if inhaled. H335: May cause respiratory irritation. Ereztech P3553 Page 1 of 13 Revision: 1.00 Date of Issue: 11/27/2020 Phosphorus trifluoride Safety Data Sheet Section 2. Hazards Identification Precautionary Statements Prevention: P260: Do not breathe fumes/gases/mists/vapors/sprays. P264: Wash skin thoroughly after handling. P270: Do not eat, drink or smoke when using this product. P271: Use only outdoors or in a well-ventilated area. P280: Wear protective gloves/ protective clothing/ eye protection/ face protection. -

Safety Data Sheet Modified: 2016-02-12 1: Identification of Substance / Mixture 1
According to Regulation (EC) No. 1272/2008 Revision: 1.06 Language : English Safety Data Sheet Modified: 2016-02-12 1: Identification of substance / mixture 1. Product Identifier Substance Product Name Silver hexafluorophosphate Product Code 002864 CAS Number 26042-63-7 Other Names IUPAC MFCD Number MFCD00003415 EC/EINECS 247-428-6 REACH Number 2. Relevant identified uses of the substance or mixture and uses advised against Research and Development 3. Details of the supplier of the safety data sheet Fluorochem Ltd Unit 14,Graphite Way Hadfield Derbyshire Telephone: +44(0)1457 860111 SK13 1QH Fax: +44(0)1457 892799 UK Email: [email protected] 4. Emergency telephone number +44(0)7855 268577 - 2. Hazards Identification 1. Classification of the substance or mixture H314 Skin Corr. 1B R34, R23/34/35 H318 Eye Dam. 1 R41 H400 Aquatic Acute 1 R50, R50/53 No Resource File 2. Label elements Signal Word Danger Hazard Statements H314 Causes severe skin burns and eye damage. H318 Causes serious eye damage. H400 Very toxic to aquatic life. Precautionary Phrases P273 Avoid release to the environment. P301 + P330 IF SWALLOWED: rinse mouth. Do NOT induce vomiting. + P331 P303 + P361 IF ON SKIN (or hair): Remove/Take off immediately all contaminated clothing. Rinse skin with + P353 water/shower. P304 + P340 IF INHALED: Remove victim to fresh air and keep at rest in a position comfortable for breathing. P305 + P351 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and + P338 easy to do. Continue rinsing. P501 Dispose of contents/container to local regulations 3. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub
US 20050044778A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub. Date: Mar. 3, 2005 (54) FUEL COMPOSITIONS EMPLOYING Publication Classification CATALYST COMBUSTION STRUCTURE (51) Int. CI.' ........ C10L 1/28; C1OL 1/24; C1OL 1/18; (76) Inventor: William C. Orr, Denver, CO (US) C1OL 1/12; C1OL 1/26 Correspondence Address: (52) U.S. Cl. ................. 44/320; 44/435; 44/378; 44/388; HOGAN & HARTSON LLP 44/385; 44/444; 44/443 ONE TABOR CENTER, SUITE 1500 1200 SEVENTEENTH ST DENVER, CO 80202 (US) (57) ABSTRACT (21) Appl. No.: 10/722,127 Metallic vapor phase fuel compositions relating to a broad (22) Filed: Nov. 24, 2003 Spectrum of pollution reducing, improved combustion per Related U.S. Application Data formance, and enhanced Stability fuel compositions for use in jet, aviation, turbine, diesel, gasoline, and other combus (63) Continuation-in-part of application No. 08/986,891, tion applications include co-combustion agents preferably filed on Dec. 8, 1997, now Pat. No. 6,652,608. including trimethoxymethylsilane. Patent Application Publication Mar. 3, 2005 US 2005/0044778A1 FIGURE 1 CALCULATING BUNSEN BURNER LAMINAR FLAME VELOCITY (LFV) OR BURNING VELOCITY (BV) CONVENTIONAL FLAME LUMINOUS FLAME Method For Calculating Bunsen Burner Laminar Flame Velocity (LHV) or Burning Velocity Requires Inside Laminar Cone Angle (0) and The Gas Velocity (Vg). LFV = A, SIN 2 x VG US 2005/0044778A1 Mar. 3, 2005 FUEL COMPOSITIONS EMPLOYING CATALYST Chart of Elements (CAS version), and mixture, wherein said COMBUSTION STRUCTURE element or derivative compound, is combustible, and option 0001) The present invention is a CIP of my U.S. -

Alphabetical Index of Substances and Articles
ALPHABETICAL INDEX OF SUBSTANCES AND ARTICLES - 355 - NOTES TO THE INDEX 1. This index is an alphabetical list of the substances and articles which are listed in numerical order in the Dangerous Goods List in Chapter 3.2. 2. For the purpose of determining the alphabetical order the following information has been ignored even when it forms part of the proper shipping name: numbers; Greek letters; the abbreviations “sec” and “tert”; and the letters “N” (nitrogen), “n” (normal), “o” (ortho) “m” (meta), “p” (para) and “N.O.S.” (not otherwise specified). 3. The name of a substance or article in block capital letters indicates a proper shipping name. 4. The name of a substance or article in block capital letters followed by the word “see” indicates an alternative proper shipping name or part of a proper shipping name (except for PCBs). 5. An entry in lower case letters followed by the word “see” indicates that the entry is not a proper shipping name; it is a synonym. 6. Where an entry is partly in block capital letters and partly in lower case letters, the latter part is considered not to be part of the proper shipping name. 7. A proper shipping name may be used in the singular or plural, as appropriate, for the purposes of documentation and package marking. - 356 - INDEX Name and description Class UN No. Name and description Class UN No. Accumulators, electric, see 4.3 3292 Acid mixture, nitrating acid, see 8 1796 8 2794 8 2795 Acid mixture, spent, nitrating acid, see 8 1826 8 2800 8 3028 Acraldehyde, inhibited, see 6.1 1092 ACETAL 3 1088 -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -
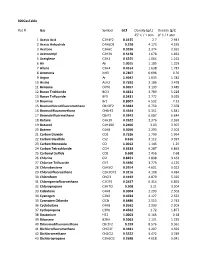
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Phosphorus Compounds
Phosphorus Compounds Table of Contents How to Cite This Article Phosphorus Compounds, Compounds of phosphorus play vital roles in the metabolism of both plants and animals. The energy "currency" of all living things, adenosine triphosphate (ATP), is probably the best-known organic phosphorus compound, but others are constituents of nervous tissue, cell membranes, and other tissues, as well as of many coenzymes. Phosphates also are key components of DNA and RNA, which carry genetic information in all organisms. Many phosphorus compounds are used in industrial processes. Most phosphorus compounds exhibit covalent properties. Ionic compounds, such as sodium 3− phosphide (Na3P), are few because the formation of P ions from P atoms requires a considerable amount of energy. Similarly, P5+ ions do not exist due to the very high ionization energies involved. The chemistry of phosphorus in forming compounds generally is similar to that of carbon and nitrogen. Phosphine Phosphine, PH3, is a very toxic gas with a garliclike odor. When inhaled it can cause restlessness, followed by tremors, fatigue, drowsiness, nausea, and often severe gastric pain. In most cases recovery occurs without aftereffects, but some cases may result in coma or convulsions and even death. Phosphine can be readily prepared by reacting calcium or aluminum phosphide with dilute acid. On a large scale, PH3 is obtained by action of sodium hydroxide on white phosphorus; − hypophosphite (H2PO 2) is also formed. Pure phosphine is not spontaneously flammable. It is readily oxidized by air when ignited, and explosive mixtures may be formed. Phosphine is sparingly soluble in water, and an aqueous solution of PH3 is neither acidic nor basic. -

United States Patent Office Patented June 15, 1971
3,584,999 United States Patent Office Patented June 15, 1971 Alternatively, the POF may be pumped into a reactor 3,584,999 containing the hydrogen fluoride and the reaction carried MANUFACTURE OF PHOSPHORUS out at 10-100° C. or higher. PENTAFLUORIDE Robert A. Wiesboeck, Atlanta, Ga., assignor to United The mole ratio of the reactants is not critical; however, States Steel Corporation, Pittsburgh, Pa. best results were obtained when 2-4 (preferably 3) moles No Drawing. Filed Jan. 16, 1968, Ser. No. 698,128 of hydrogen fluoride are employed per mole of phos Int, C. C01b. 25/10 phoryl fluoride. U.S. C. 23-205 10 Claims Approximately one-half of the POF is converted to PFs of high purity. If the product is removed from the O reactor at temperatures above 20° C., some hydrogen. ABSTRACT OF THE DISCLOSURE fluoride and a trace of phosphoryl fluoride are also ob Phosphoryl fluoride is reacted with hydrogen fluoride tained. These impurities can be separated by fractional to form phosphorus pentafluoride and hexafluorophos condensation at -40° C. phoric acid, and the hexafluorophosphoric acid may be The remaining liquid phase of the reaction mixture is reacted with sulfur trioxide, pyrosulfuric acid or fluoro essentially 60-65 percent hexafluorophosphoric acid. The sulfonic acid to form additional phosphorus pentafluoride. yield of phosphorus pentafluoride can be increased by The hexafluorophosphoric acid from any source may be reaction of the remaining liquid with sulfur trioxide, as reacted with the sulfur trioxide and/or pyrosulfuric acid described hereinafter. Total conversion of POF to PFs, to liberate phosphorus pentafluoride. -

Durham E-Theses
Durham E-Theses A study of transition metal compounds in liquid hydrogen chloride Symon, David Allen How to cite: Symon, David Allen (1972) A study of transition metal compounds in liquid hydrogen chloride, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/9115/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk To Tina INDEX PREFACE i ACKNOWLEDGEMENTS ii SUMMARY iii CHAPTER I INTRODUCTION 1 1.1.1 Non-Aqueous Solvents 1 1.1.2 History 2 1.1.3 Theory of the Solvent 9 1.2 Protonation of neutral transition metal complexes 14 1.2.1 History 14 CHAPTER 2 EXPERIMENTAL TECHNIQUES 18 2.1 The Vacuum System 18 2.2 Apparatus 23 2.3 Inert Atmosphere Glove Box Techniques 29 2.4 Determination of physical properties 30 2.4.1 Infrared spectra 30 2.4.2 '''H Nuclear Magnetic resonance studies 42 2.4.3 Ultra violet/visible spectra 45 2.4.4