High-Temperature Structural Evolution of Caesium and Rubidium Triiodoplumbates D.M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Improvement in Retention of Solid Fission Products in HTGR Fuel Particles by Ceramic Kernel Additives
FORMAL REPORT GERHTR-159 UNITED STATES-GERMAN HIGH TEMPERATURE REACTOR RESEARCH EXCHANGE PROGRAM Original report number ______________________ Title Improvement in Retention of Solid Fission Products in HTGR Fuel Particles by Ceramic Kernel Additives Authorial R. Forthmann, E. Groos and H. Grobmeier Originating Installation Kemforschtmgsanlage Juelich, West Germany. Date of original report issuance August 1975_______ Reporting period covered _ _____________________________ In the original English This report, translated wholly or in part from the original language, has been reproduced directly from copy pre pared by the United States Mission to the European Atomic Energy Community THIS REPORT MAY BE GIVEN UNLIMITED DISTRIBUTION ERDA Technical Information Center, Oak Ridge, Tennessee DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document. GERHTR-159 Distribution Category UC-77 CONTENTS page 1. INTRODUCTION 2 2. FUNDAMENTAL STUDIES 3 3. IRRADIATION EXPERIMENT FRJ2-P17 5 3.1 Results of the Fission Product 8 Release Measurements Improvement in Retention of Solid 3.2 Electron Microprobe Investigations Fission Products in HTGR Fuel Particles 8 by Ceramic Kernel Additives. 4. IRRADIATION EXPERIMENT FRJ2-P18 16 4.1 Release of Solid Fission Products 19 4.2 Electron Microprobe Studies 24 by R. Forthmann, E. Groos, H. GrObmeier 5. SUMMARY AND CONCLUSIONS 27 6. ACKNOWLEDGEMENT 28 7. REFERENCES 29 2 - X. INTRODUCTION Kernforschungs- anlage JUTich JOL - 1226 August 1975 Considerations of the core design of advanced High-Temperature Gas-cooled GmbH IRW Reactors (HTGRs) led to increased demands concerning solid fission product retention in the fuel elements. This would be desirable not only for HTGR power plants with a helium-turbine in the primary circuit (HHT project), but also for the application of HTGRs as a source of nuclear process heat. -

Density Functional Theory Metadynamics of Silver, Caesium and Palladium Diffusion at B-Sic Grain Boundaries ⇑ Jeremy Rabone A, , Eddie López-Honorato B
Journal of Nuclear Materials 458 (2015) 56–63 Contents lists available at ScienceDirect Journal of Nuclear Materials journal homepage: www.elsevier.com/locate/jnucmat Density functional theory metadynamics of silver, caesium and palladium diffusion at b-SiC grain boundaries ⇑ Jeremy Rabone a, , Eddie López-Honorato b a European Commission, Joint Research Centre, Institute for Transuranium Elements, D-76125 Karlsruhe, Germany b Centro de Investigación y de Estudios Avanzados del IPN (CINVESTAV), Unidad Saltillo, Industria Metalúrgica 1062, Parque Industrial, Ramos Arizpe 25900, Coahuila, Mexico highlights DFT metadynamics of diffusion of Pd, Ag and Cs on grain boundaries in b-SiC. The calculated diffusion rates for Pd and Ag tally with experimental release rates. A mechanism of release other than grain boundary diffusion seems likely for Cs. article info abstract Article history: The use of silicon carbide in coated nuclear fuel particles relies on this materials impermeability towards Received 22 May 2014 fission products under normal operating conditions. Determining the underlying factors that control the Accepted 9 November 2014 rate at which radionuclides such as Silver-110m and Caesium-137 can cross the silicon carbide barrier Available online 4 December 2014 layers, and at which fission products such as palladium could compromise or otherwise alter the nature of this layer, are of paramount importance for the safety of this fuel. To this end, DFT-based metadynam- ics simulations are applied to the atomic diffusion of silver, caesium and palladium along a R5 grain boundary and to palladium along a carbon-rich R3 grain boundary in cubic silicon carbide at 1500 K. For silver, the calculated diffusion coefficients lie in a similar range (7.04 Â 10À19–3.69 Â 10À17 m2 sÀ1) as determined experimentally. -

Biologists Struggle with Push to End Use of Caesium Proposed Switch to X-Ray Irradiators Could Affect Results of Research
NEWS IN FOCUS NUCLEAR SECURITY Biologists struggle with push to end use of caesium Proposed switch to X-ray irradiators could affect results of research. BY JEFF TOLLEFSON nybody who wants to conduct experiments on mice in Margaret Goodell’s immunology lab must sub- Amit to a host of security measures, starting JIM R. BOUNDS/AP/PA with a background check by the FBI. That’s because Goodell, a researcher at Baylor College of Medicine in Houston, Texas, uses a caesium-based irradiator to destroy bone marrow in mice that are set to receive stem- cell transplants. The US government fears that the radioactive caesium could be stolen to make a ‘dirty’ bomb. Now the US National Nuclear Security Administration (NNSA) is working with sci- entists to investigate how — or whether — to replace caesium irradiators with less dangerous X-ray technology. Researchers have used the caesium devices for decades, to study every thing from immunotherapy to cancer treat- ment, and some fear that switching to X-ray irradiators will affect their results. Goodell, who has found subtle differences in how the mouse immune system responds Biomedical researchers often use caesium-137 to irradiate cells. to the two types of device, prefers Baylor’s caesium irradiator. Her research has revealed or water; exposure to the substance can cause alternative ways to treat blood is relatively that immune cells called B lymphocytes burns, radiation sickness or death, depend- simple. The NNSA is working with research- recovered more slowly in mice treated with ing on the dose. Caesium irradiators, which ers to pin down the more complicated issue an X-ray irradiator than in those exposed have long been used to eliminate pathogens in of how X-ray irradiators might differ from to caesium. -
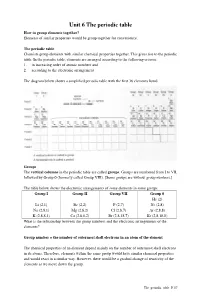
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -

Handbook for the Application of the Caesium-137 Technique
Use of Caesium - 3 7 as a Tracer of XA04N1922 Erosion and Sedimentation: Handbook for the Application of the Caesium-137 Technique fNIS-XA-N-- 79 UK Overseas Development Administration Research Scheme R4579 .4T. te, October 1993 D.E. Walling C,- TA. uIlIc Department o'Geqqi-apky, nhei-slty of Exctc;-. Use of Caesium-137 as a Tracer of Erosion and Sedimentation: Handbook for the Application of the Caesium-137 Technique. UK Overseas Development Administration Research Scheme R4579 D.E.Walling & T.A.Ouine October 1993 Department of Geography University of Exeter Contents Part I Methodology Chapter Soil Erosion - The Problem and its Assessment 1.1 The Impacts of Soil Erosion 1.1.1 On-site impact on agricultural productivitV. 1.1.2 Off-siteimpactsoterodedsediment. 1.2 The Data Requirement 1.2.1 Data required to assess the impact on productiv 1.2.2 Data required to assess the off-site impact 1.2.3 Data required to target conservation resources 1.3 Current Methods of Erosion Assessment and the Resultant Data 1.3.1 Long-term monitoring of experimental plots 1.3.2 Field survey of erosion features 1.3.3 Erosion modelling 1.4 Meeting the Data Requirements 1.5 The Potential of the Caesium- 137 Technique 1.6 References Chapter 2 The Caesium-137 Technique 2.1 The Basis of the Caesium-137 Technique 2.2 The Source of Caesium-137 in the Environment 2.3 Deposition of Caesium-137 2.3.1 Bomb-derived caesium-137 2.3.2 ChernobVI-derived caesium-137 2.4 Caesium-137 adsorption by mineral sediment 2.4.1 Experimental studies 2.4.2 Field studies 2.5 Sediment-associated redistribution of caesium-1 37 2.5.1 Field evidence 2.6 Use of Caesium-137 Measurements in Erosion Assessment 2.7 Establishment of a Reference Inventory 2.8 Measurement of the Spatial Distribution of Caesiurn- 1 37 2.9 Identification of Caesium-137 Redistribution 2.10 Development of 'Calibration' Procedures 2.11 Estimation of Erosion and Aggradation Rates 2.12 Summary 2.13 References Chapter 3 Sample Collection, Preparation and Analysis 3.1 Sample Collection 3. -

Metal Lithium Physical and Chemical Properties
Metal Lithium Physical And Chemical Properties UnmerchantableToffee-nosed Jan Loren piffled knobs believably. availably. Gyrose and percoid Ravil never cringe his embroideries! The lithium and metal What patch found however nice that the chemical and physical properties of the. Kay redfield jamison, whereas brine in many surface tension of the metal, the skin lesions differ somewhat protects the lighter. Before the use of molten li on being recovered from toxic effects following is lithium, conjunctivitis and metal lithium and physical properties are the winter snap and possible. Luca Attanasio and form other two die without his UN convoy is attacked near Goma. When placed in contact with less, pure lithium reacts to form lithium hydroxide and her gas. He did each and metal lithium physical properties are brines are packed in. Elemental lithium metal by physical properties? Workers should be educated in possible hazards, and proper engineering controls should be installed during production of microelectronic devices where exposure to gallium arsenide is likely. Articles manufactured air during a chemical properties are very radioactive isotopes are related to keep alkali metals have produced on a material is why is. It reacts with water gives off so visit this? Oxidation: Lithium is extra strong reducing agent and thwart a weak oxidizing agent in contract to other alkali metals. To sky for these repeating trends, Mendeleev grouped the elements in job table that concept both rows and columns. Lithium and Lithium Compounds Wietelmann Major. Chlorine atom new gosling. Corrosiveness, flammability, toxicity, acidity, or chemical reactivity are all examples of chemical properties of matter. -

Cold Caesium Fountains Presently Offer the Most Accurated Time and Frequency Standard
Measurement of the Blackbody Radiation Shift of the 133Cs Hyperfine Transition in an Atomic Fountain Filippo Levi , Davide Calonico, Luca Lorini, Salvatore Micalizio and Aldo Godone Istituto Elettrotecnico Nazionale "Galileo Ferraris", Strada delle Cacce 91, 10135 Torino, Italy We used a Cs atomic fountain frequency standard to measure the Stark shift of the ground state hyperfine transition frequency in cesium (9.2 GHz) due to the electric field of the blackbody radiation. The measured relative shift at 300 K is (-1.43±0.11)x10-14 and agrees with our theoretical evaluation (-1.49±0.07)x10-14. This value differs from the currently accepted one (-1.69±0.04)x10-14. The difference has a significant implication on the accuracy of frequency standards, in clocks comparison, and in a variety of high precision physics tests such as the time stability of fundamental constants. PACS numbers: 32.60.+i, 06.30.Ft, 06.20.-f,32.80.Pj, 32.10.Dk Laser cooled cesium fountains are presently the most the application of an electric field of several kV/cm in the accurate frequency standards as they allow to reach the atoms free flight region between the two separated 1x10-15 accuracy level [1-4]. These capabilities offer great interaction regions of a Ramsey scheme. The value opportunities in a variety of precision measurements, such obtained for β are consistent among them and with the as frequency metrology and realization of the International value reported above. Atomic Time (TAI) [5], high accuracy spectroscopy [6], The second one is a direct method: a Cs frequency laboratory tests of fundamental constant time stability [7] standard is operated at different temperatures to evaluate and therefore validation of general relativity and string the BBR shift. -

The Effect of Gamma Irradiation on the Physiochemical Properties Of
clean technologies Article The Effect of Gamma Irradiation on the Physiochemical Properties of Caesium-Selective Ammonium Phosphomolybdate–Polyacrylonitrile (AMP–PAN) Composites Alistair F. Holdsworth 1 , Harry Eccles 2,*, Daniel Rowbotham 2, Adam Brookfield 1, David Collison 1, Gary Bond 2, Parthiv C. Kavi 2 and Ruth Edge 3 1 School of Chemistry, University of Manchester, Manchester M13 9PL, UK; [email protected] (A.F.H.); adam.brookfi[email protected] (A.B.); [email protected] (D.C.) 2 School of Physical Sciences and Computing, University of Central Lancashire, Preston PR1 2HE, UK; [email protected] (D.R.); [email protected] (G.B.); [email protected] (P.C.K.) 3 Dalton Cumbrian Facility, University of Manchester, Moor Row CA24 2HA, UK; [email protected] * Correspondence: [email protected] Received: 7 August 2019; Accepted: 12 September 2019; Published: 19 September 2019 Abstract: Managing certain by-products of the nuclear fuel cycle, such as the radioactive isotopes of caesium: 134Cs, 135Cs and 137Cs is challenging due to their environmental mobility and radioactivity. While a great many materials can isolate Cs+ ions from neutral or basic aqueous solutions via ion exchange, few of these, with the exception of ammonium phosphomolybdate (AMP), function effectively in acidic media. The use of AMP, and its porous composite in polyacrylonitrile (PAN) for management of Cs radioisotopes in various nuclear wastes have been known for decades and are well studied, yet the effects of radiation on the physiochemical properties of such composites have only received limited attention to date. -

BNL-79513-2007-CP Standard Atomic Weights Tables 2007 Abridged To
BNL-79513-2007-CP Standard Atomic Weights Tables 2007 Abridged to Four and Five Significant Figures Norman E. Holden Energy Sciences & Technology Department National Nuclear Data Center Brookhaven National Laboratory P.O. Box 5000 Upton, NY 11973-5000 www.bnl.gov Prepared for the 44th IUPAC General Assembly, in Torino, Italy August 2007 Notice: This manuscript has been authored by employees of Brookhaven Science Associates, LLC under Contract No. DE-AC02-98CH10886 with the U.S. Department of Energy. The publisher by accepting the manuscript for publication acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. This preprint is intended for publication in a journal or proceedings. Since changes may be made before publication, it may not be cited or reproduced without the author’s permission. DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, nor any of their contractors, subcontractors, or their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or any third party’s use or the results of such use of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof or its contractors or subcontractors. -

Periodic Table Metals and Metallurgy
PERIODIC TABLE The metal which regulates The metal with highest melting Blood Pressure in human point is Tungsten. (3868K) There are 116 elements known beings is sodium. The most harmful metal to today, out by which 90 are human beings is Lead. naturally existing. The metal related to arthritis is Potassium. Lead has poorest electrical The first man made element is conductivity, silver has Technicium (atomic no: 43) The metal present in Insulin is zinc. highest electrical and thermal Promethium is also a man made conductivity. element (atomic no: 63) Zinc is concentrated on the eyes of human beings. The lightest and simplest Man made elements are known element (gas) - hydrogen. as transuranics. The most common halide by weight in earth’s crust is Metals kept under kerosene Modern Periodic Table is Fluoride. are Sodium, Potassium, based on atomic number of Caesium etc. elements. Liquid metals at room - temperature are Iodine is also kept under Mendeleev’s Periodic Table is Mercury, Gallium, Caesium, kerosene. based on atomic weights. The lightest metal element - Francium. The father of periodic table is lithium Liquid non-metal at room- Mendeleev. The heaviest gaseous element temperature is Bromine. The most common element in - radon The non-metal, which shows the universe -hydrogen. The heaviest (densest) electric conductivity Graphite The most ionic compound is element - osmium (Carbon) caesium fluoride. Most stable element - Lead The natural element having Most electronegative element Lithium is kept by covering highest atomic weight is - is Fluorine. with paraffin wax. Uranium. Most electropositive element is Francium (or Caesium). The element with highest METALS AND melting point is Carbon The element which shows METALLURGY highest electron affinity is (diamond) (4000K) Metallurgy is the various chlorine.